
| 时间:2024-12-04 |
2024年发表于《Science Translational Medicine》(IF=15.8)

一、研究背景
胃癌(Gastric cancer,GC)是全球第五大常见恶性肿瘤,5-氟尿嘧啶(5-FU)联合铂类药物作为晚期胃癌的一线化疗方案,探讨5-FU耐药的机制有重要的临床意义。氧化磷酸化(OXPHOS),是各种肿瘤类型中化疗耐药细胞的一个显著特征。Nitrilase家族成员2(NIT2)是Nitrilase超家族的成员,NITs是一种潜在的肿瘤抑制因子,可诱导细胞凋亡周期阻滞。
二、研究结果
1、NIT2作为5-FU化学敏感蛋白介导胃癌5-FU耐药
使用CRISPR-Cas9文库靶向GC细胞系MKN45中的人代谢酶,用5-FU进行筛选(图1A)。在之前未表征的5-FU敏感性增强基因中,NIT2在该分析中成为最丰富的基因之一(图1B)。在5-FU处理下,消耗NIT2增加了肿瘤细胞活力,抑制了细胞凋亡,对未处理的细胞存活影响不大(图1C和D)。NIT2过表达(图1F)增加了5-FU处理的效果。表明NIT2使GC细胞对5-FU敏感(图1G)。为了验证NIT2在体内化疗耐药中的作用,用表达shNT、shNIT2和用rNIT2 WT或rNIT2 ED重组shNIT2的MKN45或AGS细胞建立裸鼠皮下异种移植模型,与体外结果一致(图1L和M)。Kaplan-Meier生存分析显示,NIT2低表达的胃癌患者的OS率低于NIT2高表达的胃癌患者(图1Q)。总之,结果表明NIT2是一种化学敏感蛋白,可能有助于克服5-FU耐药性。
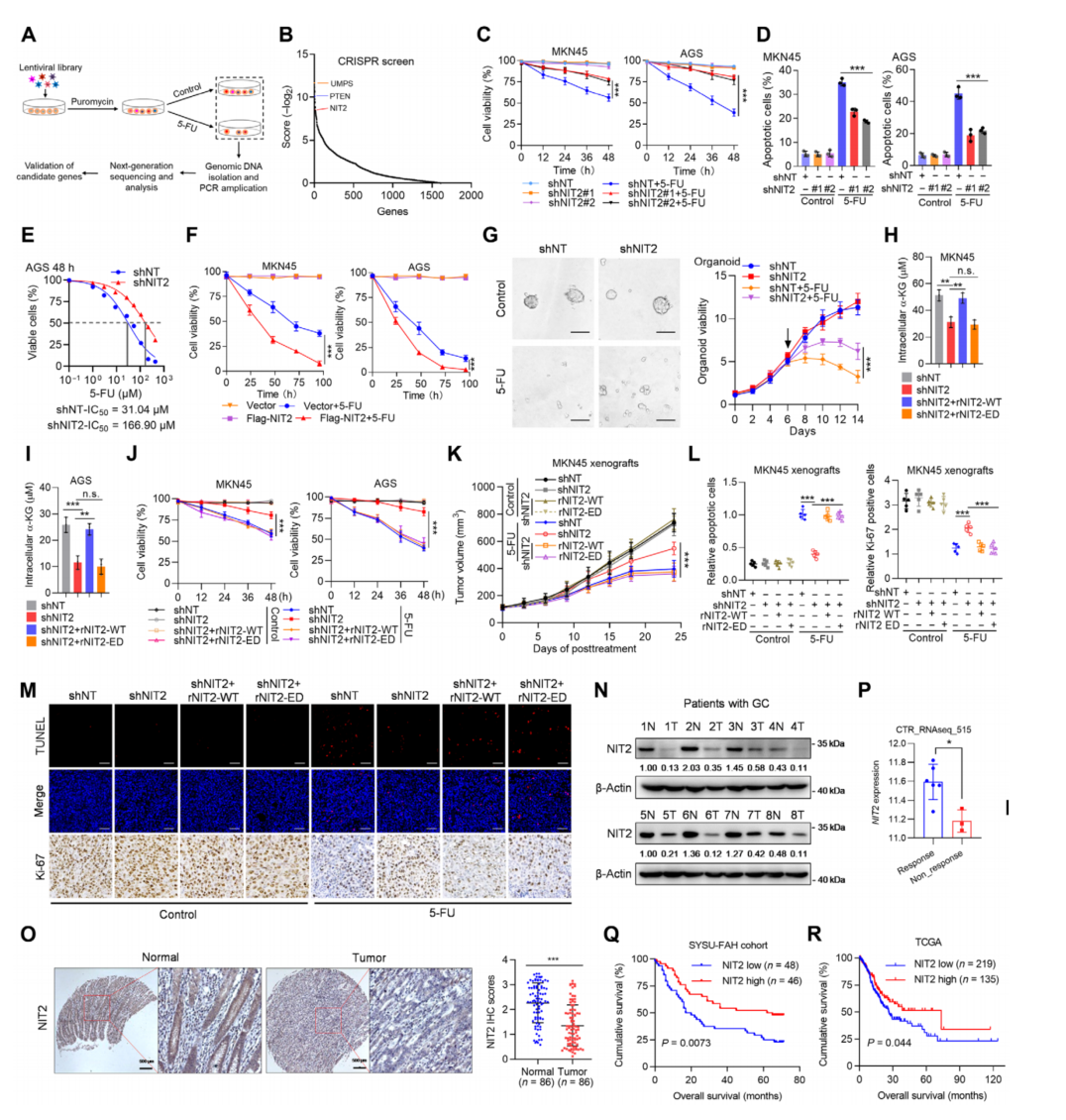
图1 NIT2作为5-FU化学敏感蛋白介导胃癌5-FU耐药
2、NIT2通过还原H3K14ac抑制OXPHOS
为研究NIT2缺失支持5-FU处理下细胞存活的机制,对暴露于5-FU下表达shNT和shNIT2的MKN45细胞进行RNA-seq分析。富集分析显示,在5-FU处理下,NIT2敲低后OXPHOS通路的上调显著富集。LC-MS分析显示,5-FU处理后,NIT2缺失细胞中二磷酸腺苷(ADP)/三磷酸腺苷(ATP)比值降低(图2B)。为了验证NIT2通过抑制OXPHOS增加5-FU敏感性,在5-FU暴露后用二甲双胍(一种OXPHOS抑制剂)处理NIT2缺失的细胞。NIT2过表达和二甲双胍处理均提高了5-FU的治疗效率,如OCR和细胞活力的降低((图2D-F)。结果表明,NIT2在5-FU处理下抑制了OXPHOS基因的表达。
用表达shNT或shNIT2的MKN45细胞在5-FU处理后进行了ATAC-seq测定。NIT2敲低显著促进了染色质的可及性,包括NDUFA13和COX20(图2I)。NIT2敲低显示H3K14ac显著增加,但在H3K9ac、H3K27ac和H3K36ac中没有增加(图2J)。为了确定NIT2缺失后H3K14ac的直接靶点,进行了ChIP-seq分析,联合RNA-seq数据分析显示,在NIT2-缺失细胞中,H3K14ac富集于NDUFA13和COX20位点(图2N、O)。这些结果表明,NIT2通过降低H3K14ac来抑制OXPHOS。
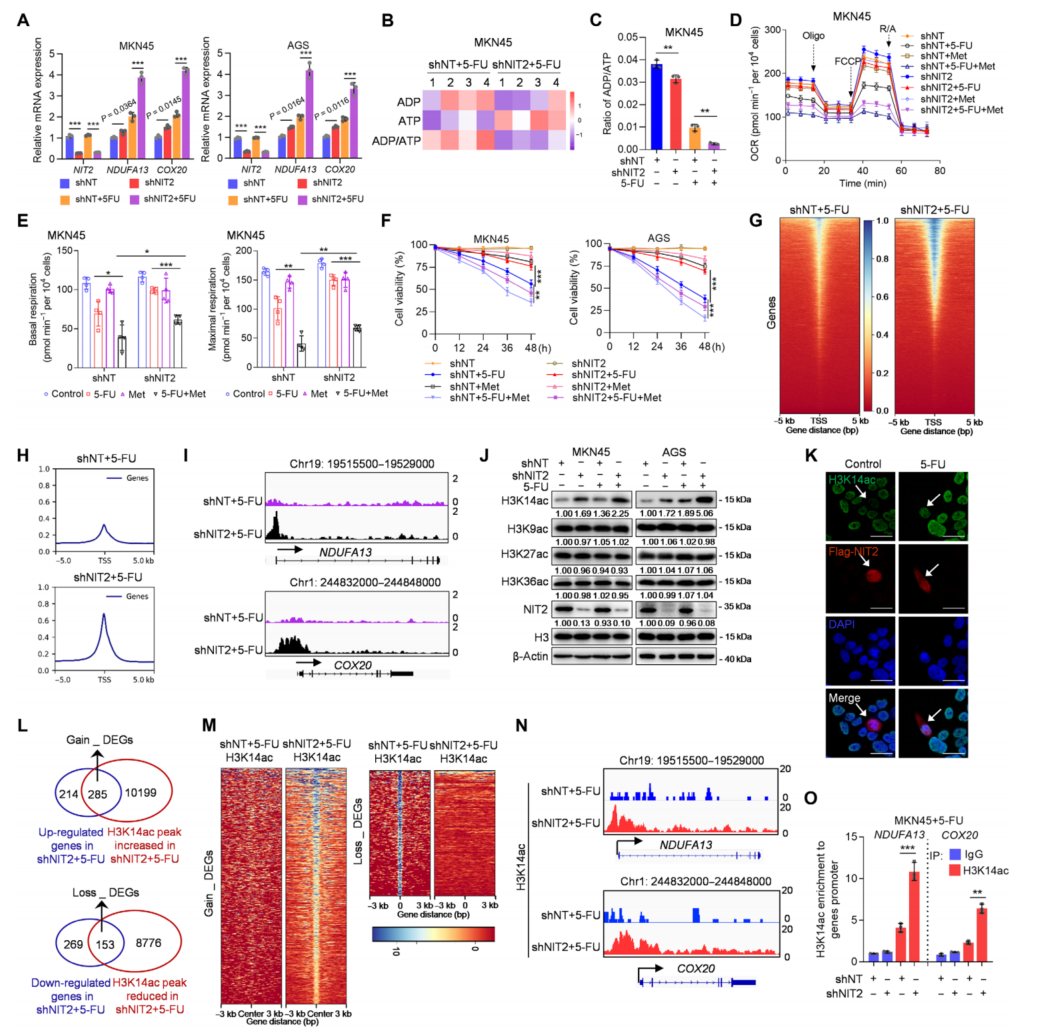
图2 NIT2通过降低H3K14ac抑制OXPHOS基因表达
3、NIT2通过减少BRD1的相分离来抑制HBO1介导的H3K14ac修饰
为了研究NIT2如何调控H3K14ac修饰,对NIT2相关蛋白进行质谱分析,并鉴定出与H3K14ac相关的BRD1。Co-IP验证了5-FU处理降低了NIT2和BRD1之间的相互作用(图3A-D)。生成了四个BRD1截断片段(图3I),结果表明BRD1的PHD结构域互作更强(图3F)。NIT2过表达抑制了BRD1与H3的结合,且抑制程度与NIT2过表达程度呈正相关(图3H),而缺乏PHD结构域的BRD1-∆PHD突变体几乎不与H3相互作用(图3G)。
对HBO1/BRD1/H3和HBO1/BRD1/H3/NIT2配合物进行了分子动力学模拟和结合自由能计算(图3I、J),表明NIT2通过改变HBO1上的H3结合域来减少HBO1和H3的结合。NIT2的结合减弱了HBO1和H3之间的相互作用。构建了增强型绿色荧光蛋白(EGFP)-BRD1质粒来检测BRD1的相分离,表明BRD1可以进行相分离(图3L)这些结果表明,当NIT2靶向HBO1复合体时,HAT活性和H3K14ac降低,从而抑制OXPHOS基因的表达,而NIT2缺失后,这种情况发生逆转(图3O)。结果表明,NIT2结合BRD1抑制HBO1复合物介导的H3K14ac修饰,从而抑制OXPHOS。
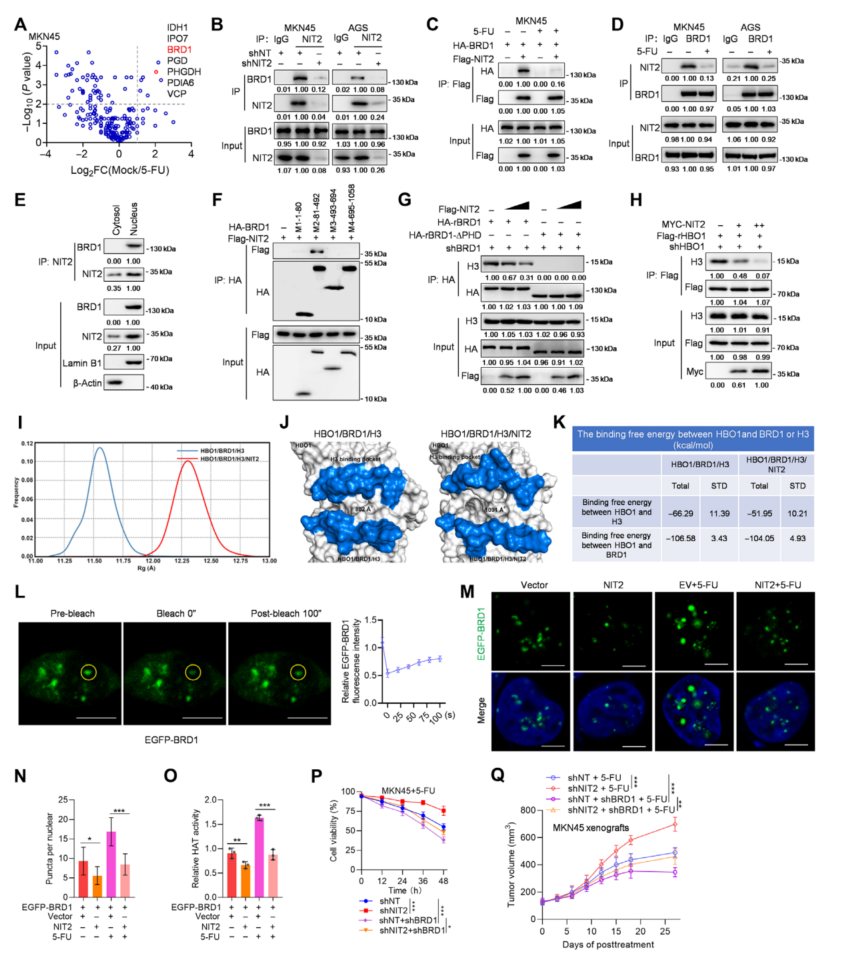
图3 NIT2通过结合BRD1抑制HBO1介导的H3K14ac修饰
4、Src介导的NIT2 Y49磷酸化将NIT2与BRD1分离,并稳定RELA
为了研究5-FU处理后NIT2与BRD1复合物解离的机制,将NIT2的磷酸化位点突变为丙氨酸(A)。Co-IP测定,5-FU处理后,NIT2 Y49A与BRD1复合物相互作用(图4B)。NIT2 Y49磷酸化(NIT2 pY49)导致BRD1与NIT2之间的相互作用的减少(图4C)。相反,NIT2 Y49A突变,模拟Y49的组成性去磷酸化,在5-FU处理下抑制EGFP-BRD1核点的形成并抑制HAT活性(图4、E和F)。5-FU诱导Src在Y49位点磷酸化NIT2(图4G-I)。
NIT2缺失增加了RELA蛋白的表达,但不影响其mRNA的表达(图4J)。5-FU处理后,细胞核中RELA的表达增加,细胞质中RELA的表达减少。与rNIT2 WT相比,5-FU处理后,rNIT2 Y49A对RELA降解的促进作用在ING4敲除后被抑制,表明RELA降解依赖于ING4(图4K)。ChIP-seq显示在5-FU处理下,NIT2 Y49A减少了RELA对NDUFA13和cox-20启动子的募集(图4M)。综上所述,表明5-FU诱导Src介导的NIT2在Y49位点磷酸化,促进其与HBO1复合物的分离,然后增强HBO1复合物与H3之间的相互作用,从而抑制ING4介导的RELA降解。

图4 Src调控的NIT2 Y49磷酸化将NIT2与BRD1分离,并稳定RELA
5、NIT2在5-FU介导的磷酸化过程中发生自噬降解
NIT2表达在5-FU耐药样本中明显较低(图5A),但mRNA表达没有改变(图5B)。5-FU处理后,蛋白酶体抑制剂MG132的刺激对NIT2蛋白的影响很小(图5D)。此外,在5-FU处理的ATG5 KO自噬缺陷细胞中,NIT2降解率明显受到抑制(图5E)。LC3是自噬的标记蛋白,发现5-FU处理后NIT2与LC3结合(图5F),增加了NIT2和LC3的共定位。为了确定负责NIT2降解的货物受体,对已知的货物受体与NIT2进行了Co-IP,结果显示只有p62与NIT2结合(图5G)。5-FU处理促进了p62与NIT2之间的相互作用(图5G)。结果表明,5-FU刺激促进了NIT2的自噬降解。5-FU处理后,NIT2 Y49A的表达得以维持(图5H),这可能是因为与NIT2 WT相比,NIT2 Y49A几乎不与LC3和p62相互作用(图5,I和J)。总之NIT2 Y49的磷酸化是NIT2蛋白不稳定的关键。
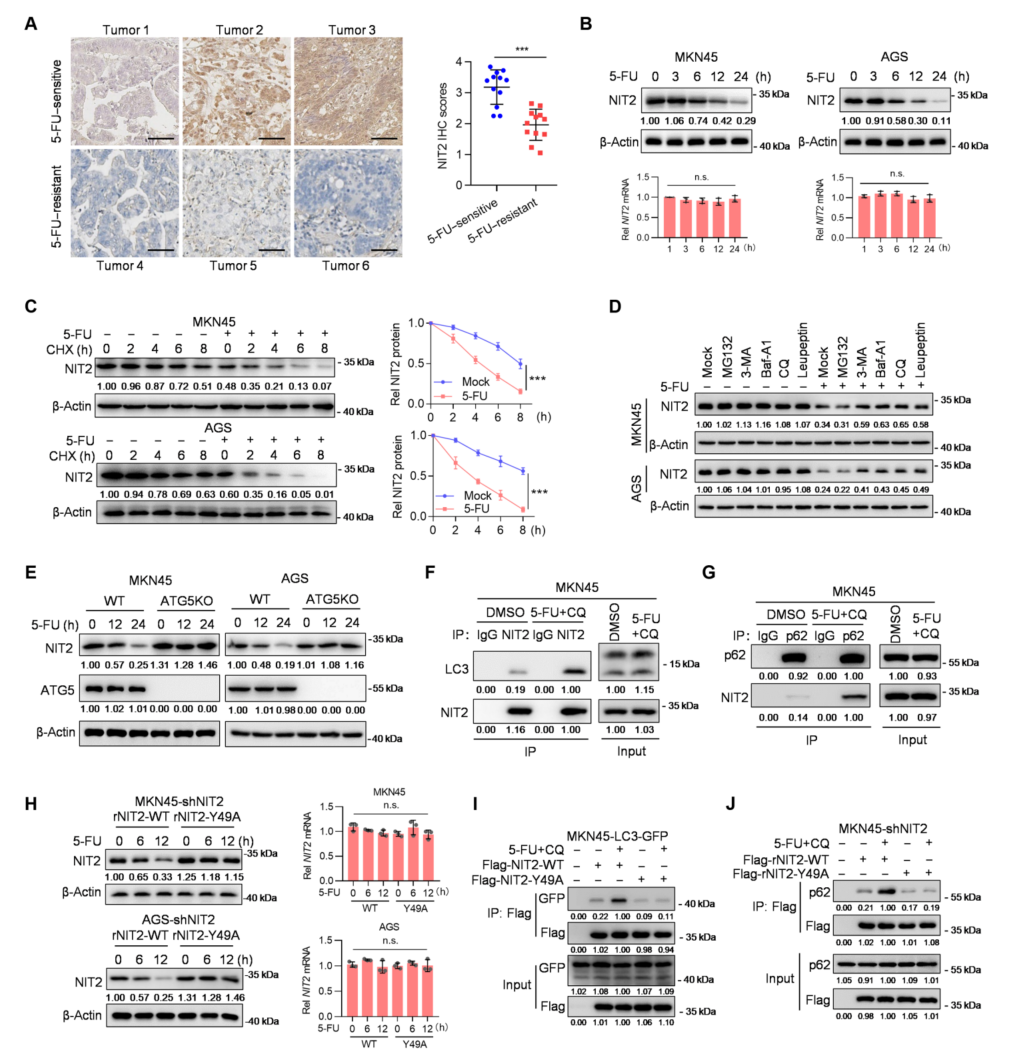
图5 5-FU诱导NIT2自噬降解
6、CCNB1IP1促进NIT2 K60残基的泛素化
由于货物受体p62识别泛素化蛋白靶点,发现在5-FU处理下,NIT2泛素化增加(图6A)。NIT2 Y49A的泛素化保持不变(图6B)。质谱分析确定5-FU调控的NIT2泛素化位点,4个潜在的泛素化残基,K52、K60、K68和K249。在5-FU处理后,只有NIT2 K60R突变体的表达几乎保持不变(图6C)。5-FU诱导了NIT2 WT的降解,但对NIT2 K60R的影响很小(图6D),而NIT2 K60R则降低了NIT2泛素化的增加(图6E)。与NIT2 WT相比,在5-FU条件下,NIT2 K60R显著抑制H3K14ac(图6H)。因此,与NIT2 WT相比,NIT2 K60R抑制OXPHOS,提高5-FU的化疗效果(图6I)。
构建了E3连接酶的CRISPR文库(图6J),发现在CCNB1IP1、ARIH1和RFWD2中,只有CCNB1IP1敲低增加了NIT2的稳定性,而在5-FU处理下,CCNB1IP1过表达降低了NIT2的丰度(图6L)。在GC患者样本中进一步证实了NIT2与CCNB1IP1蛋白表达的反向关系(图60)。这些结果表明,E3连接酶CCNB1IP1参与了5-FU诱导的K60位点NIT2泛素化。
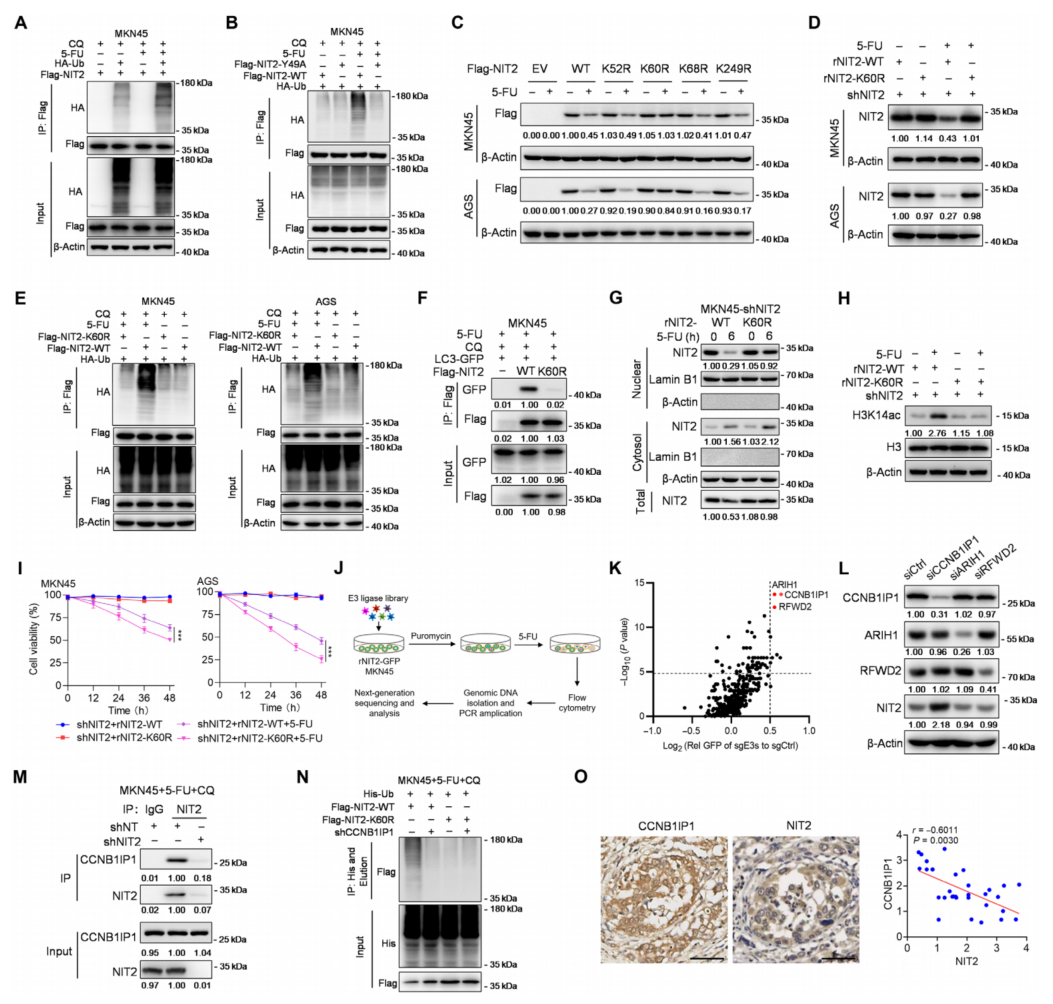
图6 在5-FU处理下,NIT2泛素化需要CCNB1IP1
7、NIT2在体内促进5-FU化疗
为了确定胃癌中NIT2与5-FU耐药性的临床相关性,发现H3K14ac、NDUFA13、COX20和CCNB1IP1在低NIT2组中高表达(图7A),与NIT2呈负相关(图7B)。在患者来源的异种移植(PDX)模型中检测了5-FU的功效(图7D)。与低NIT2表达的PDX相比,5-FU对高NIT2表达的PDX表现出更大的治疗效果,并且在低NIT2表达的PDX中,5-FU与二甲双胍联合治疗效率提高(图7E、F)。在低NIT2的PDXs中,OXPHOS基因的表达和H3K14ac的程度更高,表明对5-FU具有抗性(图7I)。与NIT2 WT相比,NIT2 Y49A促进了5-FU的治疗效果,而疗效与5-FU和二甲双胍联合治疗NIT2 WT肿瘤的疗效大致一致(图7J、K)。这些结果表明,Y49位点的NIT2磷酸化促进了体内5-FU的抗性。
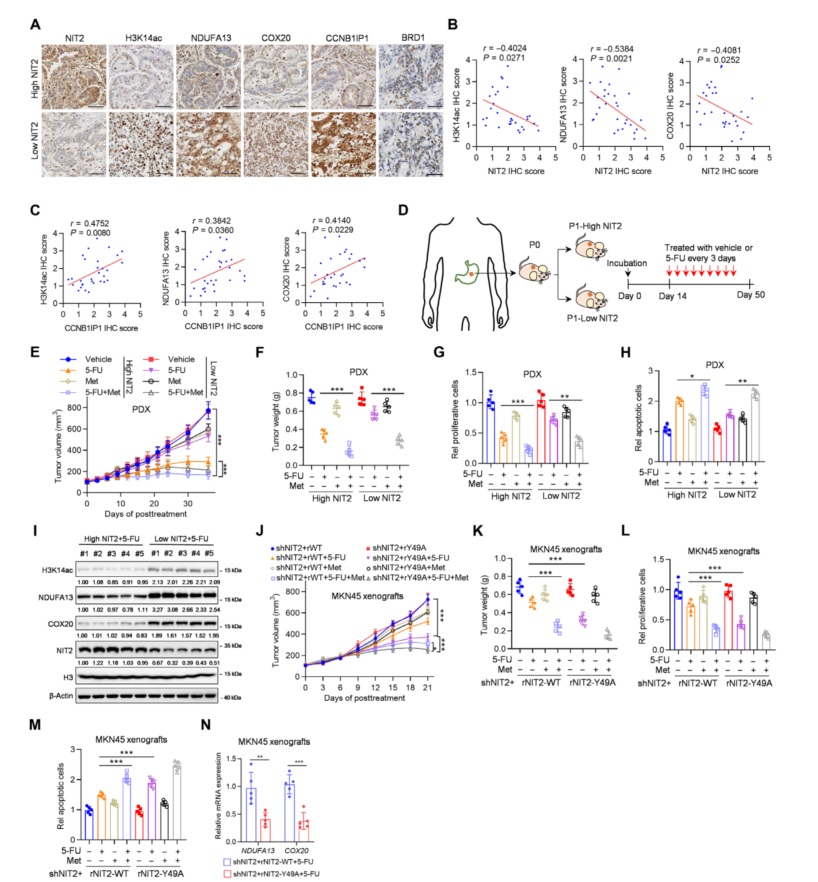
图7 NIT2在体内促进5-FU化疗
三、研究结论
NIT2的缺失或低表达导致5-FU耐药。NIT2与BRD1相互作用,抑制HBO1介导的H3K14ac乙酰化和RELA靶向氧化磷酸化(OXPHOS)基因表达。在5-FU刺激下,Src在Y49位点磷酸化NIT2,促进了NIT2与BRD1的解离,随后与E3连接酶CCNB1IP1结合,引起NIT2的自噬降解。NIT2的表达与H3K14ac和OXPHOS呈负相关,与胃癌患者的化疗反应和预后呈正相关。二甲双胍诱导的OXPHOS阻断增加了低NIT2表达的GC肿瘤患者5-FU的化学敏感性。
参考文献:
NIT2 dampens BRD1 phase separation and restrains oxidative phosphorylation to enhance chemosensitivity in gastric cancer.[J]Science Translational Medicine, 2024.