
| 时间:2024-11-15 |
2024年发表于《Circulation Research》(IF=16.5)
一、研究背景
肥厚性心肌病(HCM)是一种常见的遗传性心血管疾病,现有治疗效果并不理想。虽然基因突变被认为是HCM的主要原因;但基因治疗等最先进的技术仍无法应用于临床治疗。TFs在心脏病中的作用的机制研究仍然有限,SP1(特异性蛋白1)是第一个从哺乳动物中分离纯化的TF,SP1在心脏中的作用尚不清楚。
二、研究结果
1、心脏特异性敲除Sp1导致小鼠心肌肥大和减少舒张功能
将Sp1flox/flox小鼠与αMHC/MerCreMer(Cre+/−)小鼠杂交,有条件地删除了心肌细胞中的Sp1(图1A、B)。在他莫昔芬诱导后4周和12周,Sp1缺乏导致心肌肥厚而无心室扩张(图1C和1D)。此外,心脏形态学观察和心脏切片证实Sp1-CKO小鼠心脏增大,心室壁增厚(图1E),心体重比也显著增加(图1F)。Sp1-CKO小鼠在他莫昔芬诱导后4周无明显变化,诱导12周后,心脏舒张功能降低,收缩功能增强。
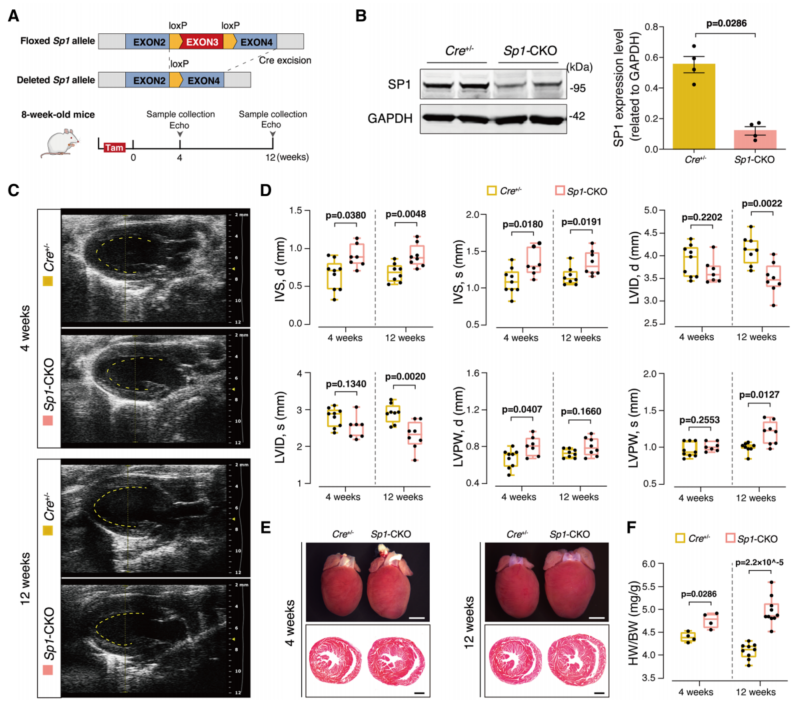
图1 Sp1缺乏导致小鼠心肌肥大
2、Sp1-CKO小鼠表现出典型的HCM组织学特征
他莫昔芬诱导后4周,心肌纤维化无明显变化;诱导后12周,Sp1-CKO小鼠与Cre+/−小鼠相比,染色显示间质纤维化严重(图2A和2B)。WGA染色显示Sp1-CKO小鼠在诱导后4周和12周心肌细胞横截面面积增大(图2C和2D)。Sp1-CKO小鼠心肌中形态异常的线粒体数量增加,这是HCM的特征(图2E);HCM标记基因ANP的表达显著增加(图2F)。注射他莫昔芬56周后Sp1-CKO小鼠的出现心肌肥厚、舒张功能受损和严重的心肌纤维化。综上所述,Sp1-CKO小鼠表现出明显的HCM组织学特征,提示Sp1在HCM发病机制中的关键作用。
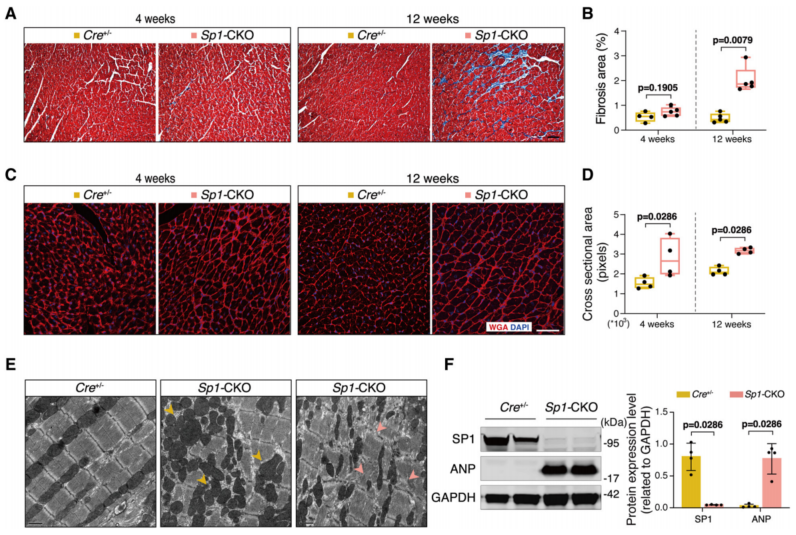
图2 心脏特异性条件敲除Sp1(特异性蛋白1)(Sp1-cko)小鼠显示肥厚性心肌病的典型组织学特征
3、SP1缺乏通过下调Tuft1表达导致HCM
为了解SP1缺陷诱导HCM的机制,对Sp1-CKO与Cre+/−的心肌细胞进行了RNA-seq。与Cre+/−小鼠相比,Sp1-CKO小鼠鉴定出333个显著差异表达基因(DEGs)(图3A)。GO分析显示,这些DEGs参与了HCM相关的生物过程(图3B)。为了确定SP1调控HCM的直接靶基因,做SP1的ChIP-seq。大于80%的结合峰富集在距离转录起始位点<1 kb的启动子区域(图3C),而Sp1-CKO小鼠转录起始位点周围SP1的结合峰和读密度显著降低(图3D)。
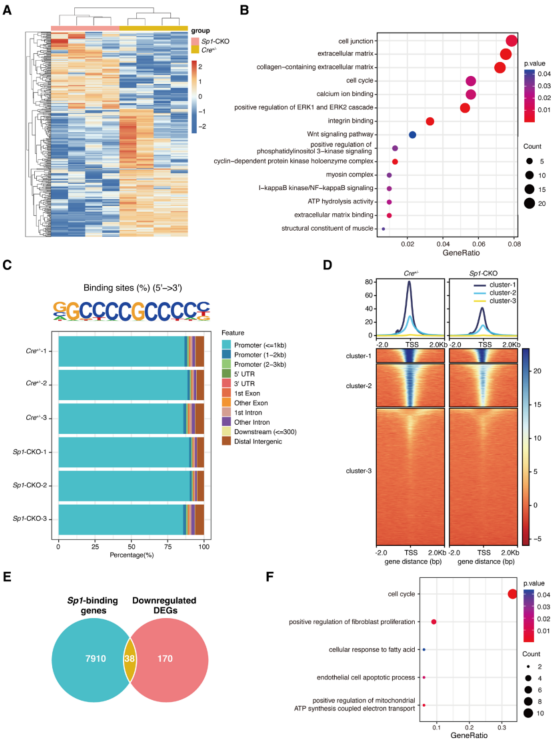
图3 Sp1(特异性蛋白1)缺乏导致小鼠肥厚性心肌病相关基因的改变
为探索Sp1缺陷诱导HCM发病机制的关键基因,将染色质免疫沉淀测序中启动子区域的Sp1结合基因与RNA测序中下调的DEGs结合,鉴定出常见基因(图3E)。GO分析显示,这些基因在HCM相关方面富集(图3F)。在Sp1缺陷心肌细胞中,Tuft1是下调最显著的基因(图4A)。ChIP-qPCR显示SP1与Tuft1启动子区直接结合(图4C)。Sp1-CKO小鼠与Cre+/−小鼠相比,Tuft1基因启动子区SP1结合峰明显降低(图4D),提示SP1缺失减少了其与Tuft1启动子的结合,导致Tuft1表达降低。数据表明,Tuft1是SP1缺陷诱导HCM的关键基因。hiPSC-CMs中SP1缺陷导致了类似于HCM心肌细胞的细胞表型(图4E、F)。在hiPSC-CMs中,TUFT1的过表达减弱了Sp1敲低诱导的细胞表型(图4E和4F),进一步验证了TUFT1是Sp1在HCM中作用的关键基因。
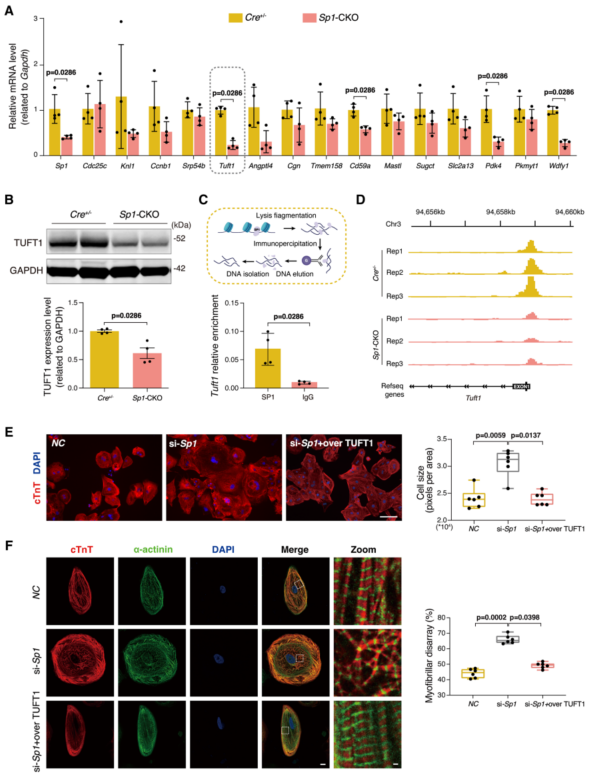
图4 Tuft1是Sp1缺陷诱导肥厚性心肌病(HCM)发病的关键基因
构建了心肌细胞特异性腺相关病毒(AAV)来探索Tuft1敲低对心脏的影响;构建了AAV-TUFT1-OE心肌细胞特异性TUFT1过表达病毒,在体内测试TUFT1过表达对心脏的影响(图5A)。过表达TUFT1可显著降低Sp1-CKO小鼠心肌肥厚和心肌细胞大小(图5C-5G)。总的来说,Tuft1是Sp1缺陷相关HCM的重要下游靶分子。
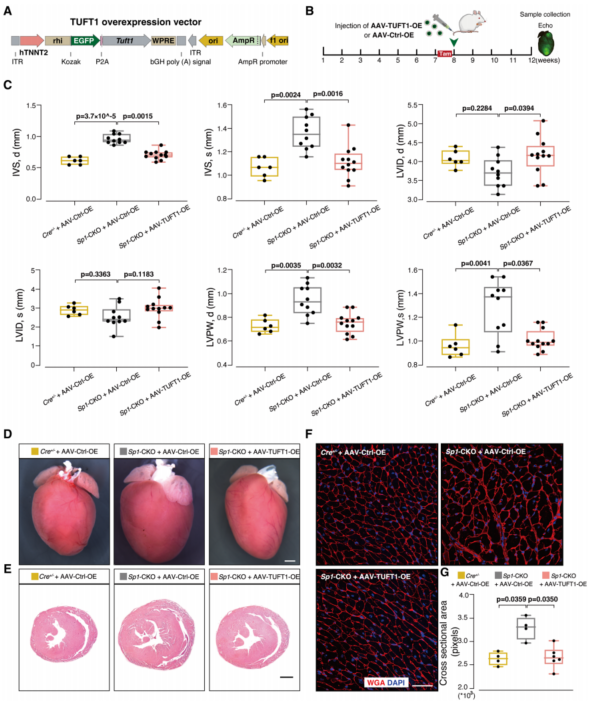
图5 TUFT1过表达可减轻小鼠SP1缺乏引起的心肌肥大
4、SP1过表达抑制R404Q小鼠HCM的进展
为了探究SP1是否可以作为HCM的干预靶点,用了携带R404Q突变的转基因小鼠模型(图6A)。AAV-SP1-OE或AAV-Ctrl-OE送入14周龄R404Q小鼠(图6B)。结果显示,与WT小鼠相比,R404Q小鼠表现出心室壁增厚、心功能异常等HCM特征,与文献报道一致(图6C)。SP1过表达病毒处理的R404Q小鼠心肌肥厚等症状减轻(图6C)。SP1过表达病毒处理的R404Q小鼠显示出正常的心脏大小、心脏与体重比和心脏结构(图6D-6F);SP1过表达病毒处理组的纤维化明显减少(图6G和6H)总之,SP1的过表达抑制了HCM小鼠的疾病进展。
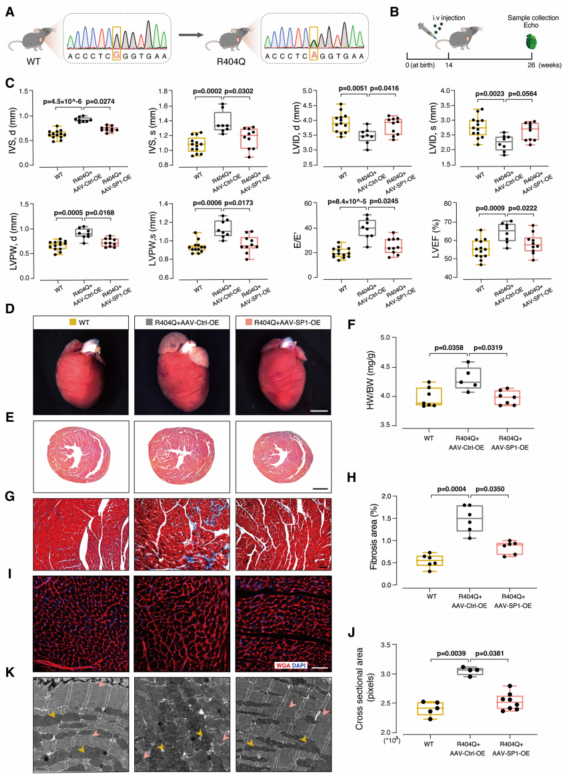
图6 SP1过表达抑制Myh6 R404Q/+ (R404Q)突变等位基因小鼠HCM的进展
5、SP1过表达逆转改变R404Q小鼠中与HCM发病机制相关基因的表达
对R404Q和WT小鼠分离的心肌细胞进行了RNA-seq,两者有不同的转录组谱(图7A)。GO分析表明,DEGs的富集与HCM密切相关(图7B)。在HCM相关GO中关注了59个改变的基因,以检测SP1过表达后它们的表达变化(图7B)。在SP1过表达后,R404Q小鼠中与细胞外基质(Apoe、Ccn4、Clec3b和Dcn)等相关的13个基因的表达改变得到纠正(图7C)。在R404Q小鼠心脏分离的心肌细胞中,Tuft1的表达显著降低,SP1过表达后也发生逆转。这些数据支持SP1过表达导致的治疗结果。总之SP1在R404Q小鼠中的过表达,可以通过逆转与HCM发病相关的异常转录组有效地抑制疾病进展。
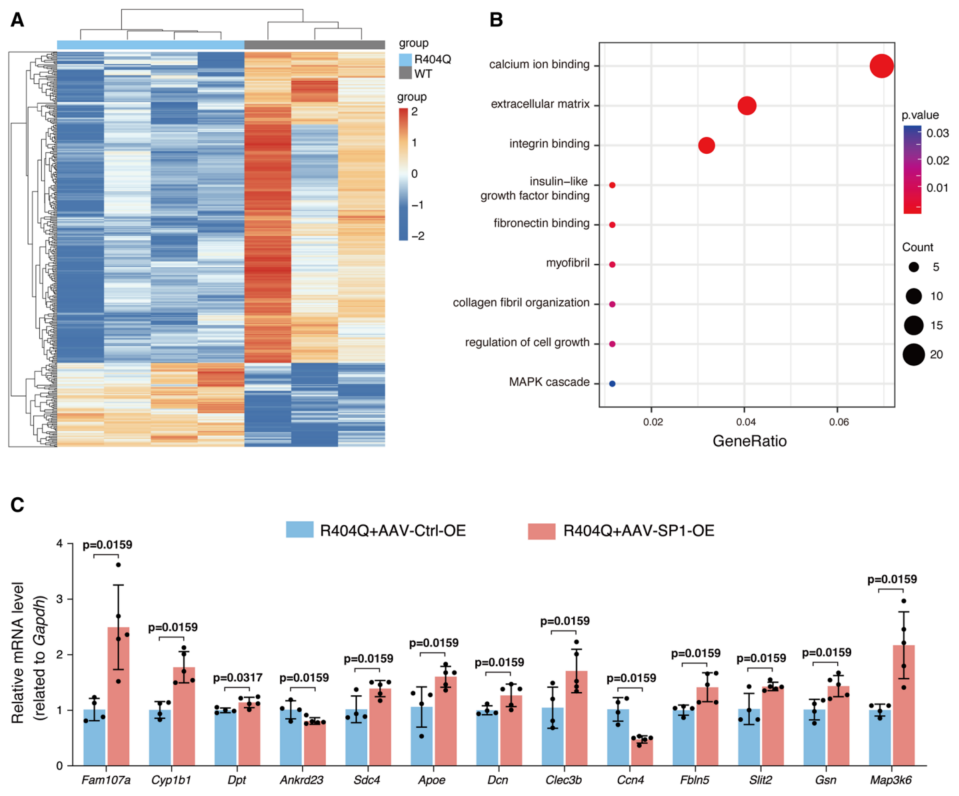
图7 SP1过表达逆转了Myh6 R404Q/+ (R404Q)小鼠突变等位基因中肥厚性心肌病相关基因的表达改变
6、针对SP1的干预逆转了HCM患者的hiPSC-CMs的细胞表型
为探索SP1是否可能是人类HCM干预的潜在靶点,研究了SP1对HCM hiPSC-CMs的影响,是一种体外HCM疾病模型(图8A)。HCM hiPSC-CMs的细胞面积较对照hiPSC-CMs明显增加、肌原纤维紊乱增加(图8B、C)。SP1过表达显著减少了HCM hiPSC-CMs中的细胞面积和肌纤维紊乱的发生率,这表明SP1过表达也可以挽救人类HCM的细胞表型(图8D和8E)。此外,TUFT1的过表达也显著减少了HCM hiPSC-CMs的细胞面积和肌纤维紊乱的发生率,提示TUFT1在HCM干预中的潜在作用。

图8 SP1过表达可减弱HCM患者的hiPSC-CMs的细胞表型
三、研究结论
小鼠的心脏特异性敲除Sp1产生典型的HCM表型,Tuft1是SP1的关键靶基因。在hiPSC-CMs和小鼠中,TUFT1过表达可以恢复Sp1敲除诱导的肥厚表型;SP1过表达可逆转HCM的肥厚表型。SP1缺乏导致HCM,SP1可能是HCM的潜在干预靶点。
参考文献:
Deficiency of Transcription Factor Sp1 Contributes to Hypertrophic Cardiomyopathy.[J]Circulation Research, 2024.