
| 时间:2024-10-18 |
2023年发表于《Journal for Immunotherapy of Cancer》(IF=10.3)
一、研究背景
免疫检查点抑制剂(ICIs)主要包括PD-1、PD-L1和CTLA-4。SETD2是唯一的组蛋白甲基转移酶,通过H3K36me3发挥关键作用。SETD2基因突变在约5%的人类癌症中普遍存在,并促进肿瘤的进展。目前还没有开发出专门针对SETD2缺陷肿瘤并显示出治疗潜力的小分子或抗体。TCGA数据集数据分析表明,SETD2突变可能与ICI免疫治疗的疗效相关,而SETD2失活的功能重要性以及如何调节免疫治疗反应仍不清楚。
二、研究结果
1、在同基因小鼠模型中敲除Setd2,使肿瘤对ICIs免疫治疗敏感
SETD2编码一种组蛋白H3K36三甲基转移酶,建立了一种同源小鼠胰腺腺癌细胞系,即KC细胞,利用CRISPR/Cas9构建Setd2-敲除(KO)KC细胞(图1A),使用抗PD-L1抗体作为ICI。与对照相比,抗PD-L1治疗显著降低了KC-sgSetd2肿瘤的生长,但对KC-对照肿瘤没有影响(图1B-E)。结果表明,Setd2敲除可使胰腺腺癌对抗PD-L1免疫治疗增敏。在小鼠黑色素瘤细胞系(B16F10)进了了进一步验证(图1F),敲低Setd2并没有明显减缓细胞增殖,但对免疫正常小鼠的抗PD-L1治疗效果有显著影响(图1G、H和I)。抗PD-L1治疗对对照B16F10肿瘤无效(图1I、J)。表明肿瘤中Setd2失活使KC胰腺腺癌和B16F10黑色素瘤对免疫检查点阻断敏感。
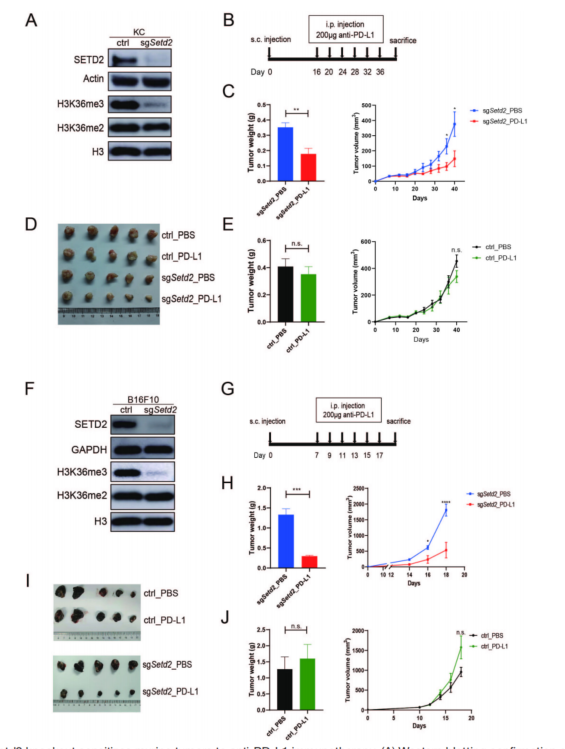
图1 Setd2敲除使小鼠肿瘤对抗pd-l1免疫治疗敏感
2、SETD2失活通过上调Stat1来激活IFNγ通路
对KC-KO/KC-WT同种异体移植肿瘤和KC-sgSetd2/ KC对照细胞进行了RNA-seq。GSEA显示,在KC-KO肿瘤和KC-sgSetd2细胞中,调节免疫反应、细胞因子产生、干扰素-γ(IFNγ)反应、抗原加工和递呈的基因集均显著上调(图2A)。IFNγ通路在肿瘤对免疫治疗的反应中起着关键作用,IFNγ通路中的关键转录因子STAT1在KC-KO肿瘤和KC-sgSetd2细胞中被显著上调和激活(图2B、C);western blotting和RT-qPCR进行了证实(图2D,E)。在体外(图2F、G)和体内(图2H)敲低SETD2,上调IFNγ诱导的趋化因子基因Cxcl9、Cxcl10和Cxcl11。这些结果表明Setd2失活增加STAT1的表达和激活,进而激活IFNγ通路,从而促进免疫相关基因的表达和免疫相关通路的激活。
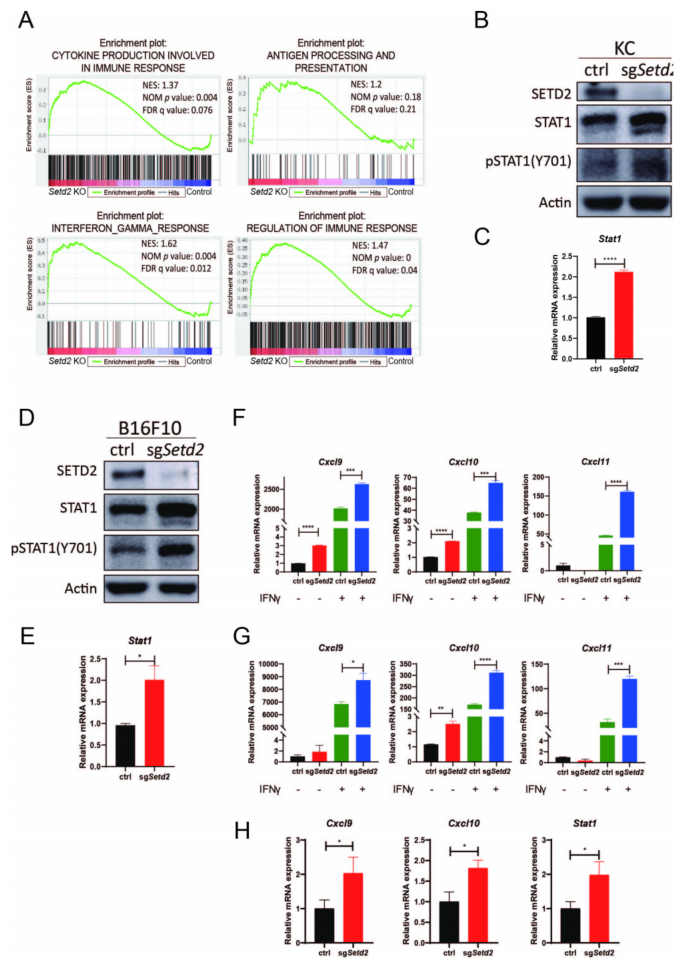
图2 Setd2失活诱导了STAT1的表达和激活
3、SETD2的缺失促进了PD-L1的表达和抗原呈递
IFNγ-JAK-STAT1轴是PD-L1表达和MHC-I抗原加工和递呈的重要途径,发现在Setd2缺失模型中,IFNγ-STAT1通路激活后PD-L1表达和抗原呈递增强。RT-qPCR证实了Pdl1的上调(图3A、B)。流式细胞术分析显示,细胞膜上PD-L1蛋白表达增加(图3C、D)。综合GSEA结果,发现Setd2缺失促进了MHC-I抗原的加工和提呈。在Setd2-KO KC细胞中组成性表达OVA,证实了sgSetd2-OVA细胞表面SIINFEKL-H-2Kb水平上调(图3G)。体外和体内实验均表明,Setd2缺乏使KC细胞对CD8+ OT-1细胞介导的杀伤效应变得敏感(图3H)。综合结果可知,Setd2缺陷通过上调Stat1激活IFNγ通路,并进一步增加Pdl1表达、IFNγ诱导的趋化因子基因表达和MHC-I抗原呈递,从而协同提高抗PD-L1免疫治疗的效果。
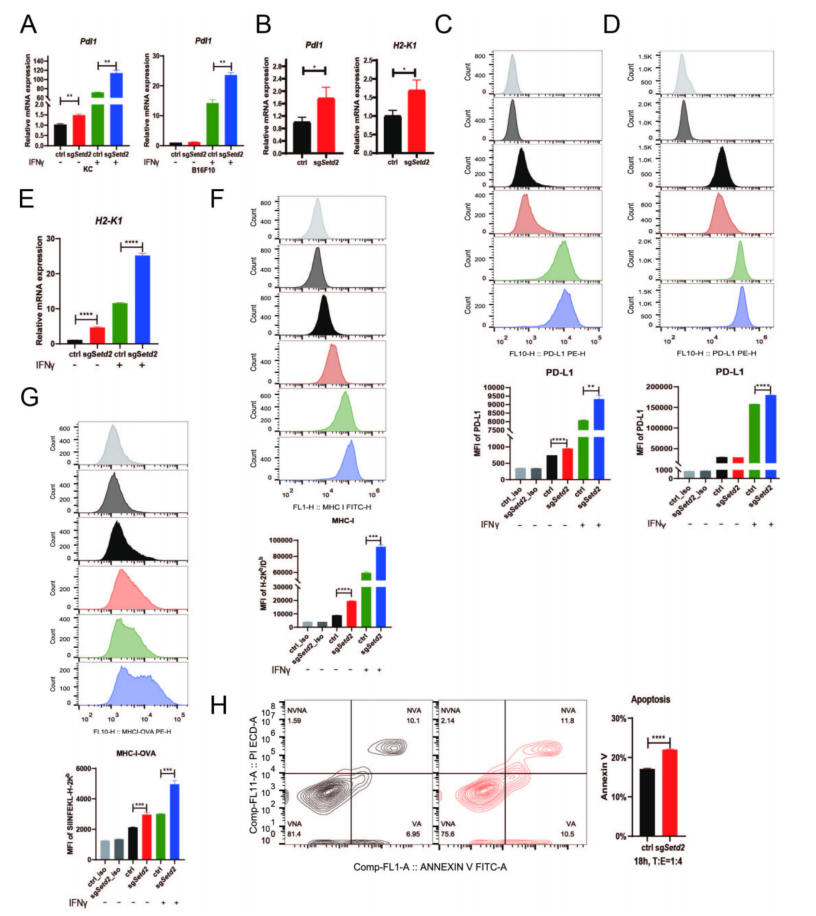
图3 Setd2失活促进IFNγ-stat1诱导的PD-L1表达和MHC-I抗原呈递
4、SETD2失活通过下调Nr2f1来促进Stat1的表达
SETD2是唯一负责H3K36me3三甲基化的酶,KC-KO和KC-WT中进行ChIP-seq看H3K36me3在细胞中的分布。KC-ctrl和KC-sgSetd2细胞中ATAC-seq表明,SETD2的缺失导致染色质可及性的全基因组改变。Nr2f1是下调最显著的转录因子,在Setd2失活细胞中该基因位置的H3K36me3富集和染色质可及性呈一致的下降趋势(图4A、B)。将NR2F1慢病毒转导至Setd2敲除细胞和对照细胞(图4C),Stat1 mRNA水平和蛋白水平均降低(图4D)。构建Flag-Nr2f1的ChIP-qPCR,证实了NR2F1与Stat1启动子结合(图4F)。GFP实验也得到了相似结果(图4G,H)。结果表明,Setd2缺失通过减少NR2F1基因体上的H3K36me3沉积和减少NR2F1启动子区域近端的染色质可及性来破坏NR2F1的转录,从而进一步以负反馈的方式增加Stat1的表达和激活。
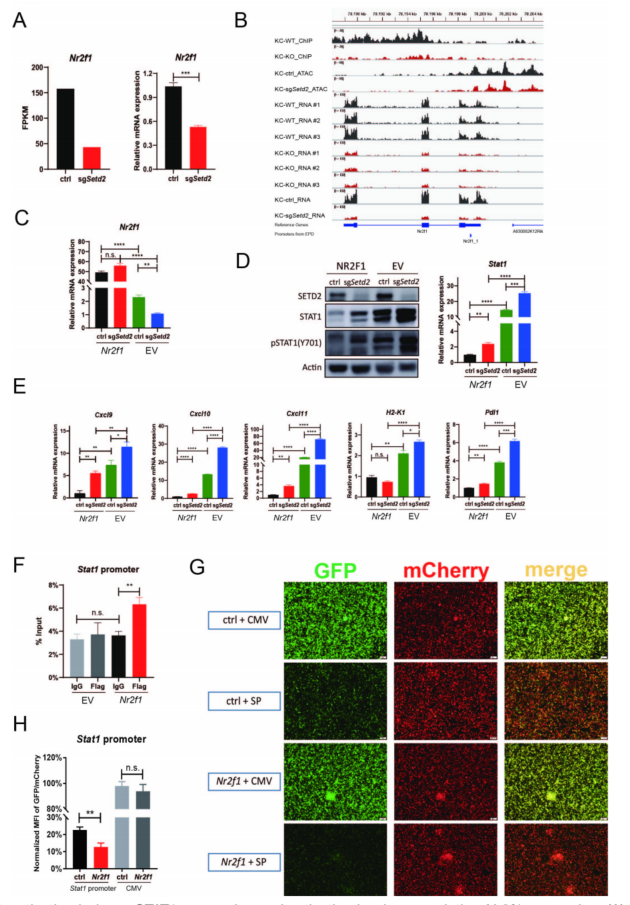
图4 Setd2失活通过下调Nr2f1的表达来诱导STAT1的表达和激活
5、SETD2缺失可重新编程肿瘤免疫微环境
对KC-KO(Setd2)和KC-WT KC细胞进行RNA-seq,ssGSEA和ESTIMATE分析显示KC-KO肿瘤的免疫细胞浸润明显高于KC-WT肿瘤(图5A)。scRNA-seq分析显示,Setd2缺陷肿瘤中浸润的T细胞、NK等细胞比例显著增加,而浸润的巨噬细胞比例显著降低(图5C、D)。M2样巨噬细胞的比例显著减少(图5E、F)。这些结果表明,Setd2缺失通过减少M2巨噬细胞浸润和增加效应T细胞浸润来炎症肿瘤微环境(TME)。轨迹分析表明,在接受抗PD-L1免疫治疗的KC-sgSetd2肿瘤中,细胞毒性CD8+ T细胞的比例显著增加(图5I、J);CD3+ T细胞、CD4+ T细胞和CD8+ T细胞的浸润增加(图5K、L)。这些发现在人类TCGA透明细胞肾细胞癌(ccRCC)队列中得到了验证。Setd2缺失导致的TME炎症有助于抗PD-L1治疗,并且细胞毒性CD8+ T细胞在免疫治疗中发挥主要作用。
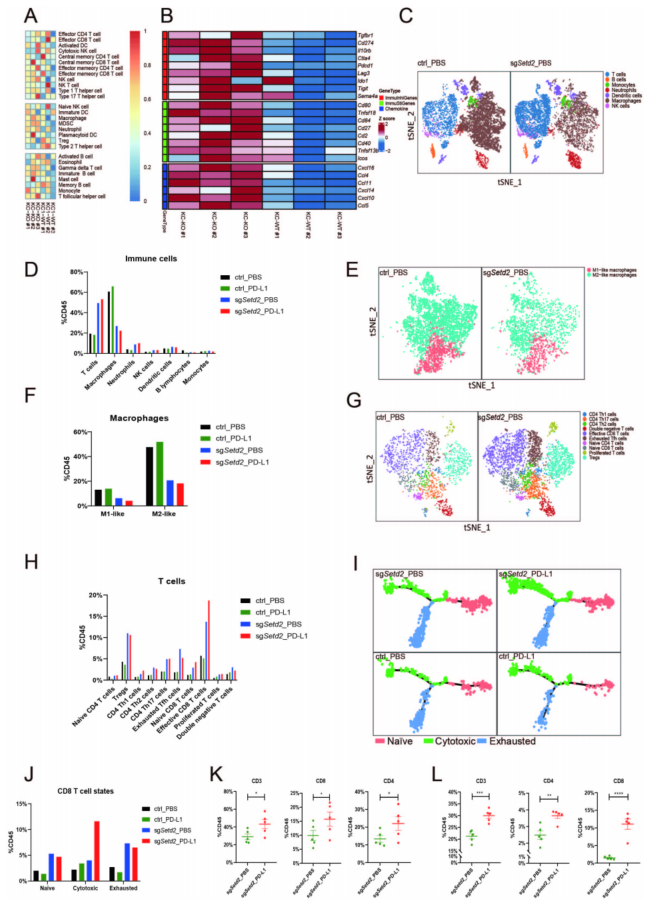
图5 肿瘤内免疫浸润的bulk RNA-seq和scRNA-seq
6、人类癌症中的SETD2-NR2F1-STAT1轴
为证实SETD2失活对STAT1的抑制依赖于NR2F1,在胰腺导管腺癌细胞系YAPC用sgRNA敲除SETD2(图6A)。STAT1在mRNA和蛋白水平上调,STAT1激活介导的CXCL10和PDL1也上调(图6A、B)。转录组分结果显示,YAPC-sgSetd2与KC-sgSetd2细胞基因特征相似(图6C、D)。ICGC胰腺癌队列的公共数据集分析显示SETD2与STAT1表达,NR2F1与STAT1表达,SETD2与PDL1表达呈负相关(图6E、F)。非配对比较显示,在SETD2高表达的肿瘤中,STAT1和PDL1的表达较低(图6G),而在NR2F1低表达的肿瘤中,STAT1的表达显著升高(图6G)。最后,对75例胰腺导管腺癌患者进行了IHC,证实了SETD2和NR2F1的表达显著正相关(图6H、I)。
NR2F1与STAT1、SETD2以及免疫抑制基因CTLA-4、IDO1、HAVCR2、LAG3的表达呈负相关(图6J),SETD2与NR2F1的表达呈正相关(图6K)。SETD2-高的肿瘤中NR2F1 mRNA水平明显高于SETD2-低和SETD2-缺失的肿瘤(图6L)。这些结果表明SETD2-NR2F1-STAT1调控的机制在人类癌细胞系和患者样本中是高度保守的,突出了SETD2缺乏在ICIs免疫治疗人类癌症疗效中的作用。
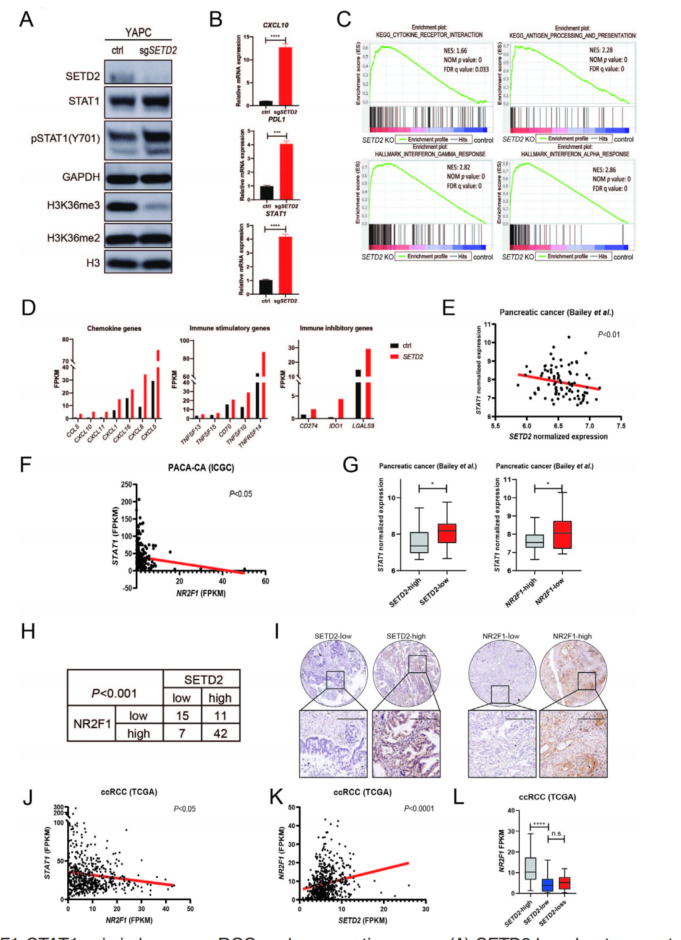
图6 SETD2-NR2F1-STAT1轴在人ccRCC和胰腺癌中的作用
7、携带SETD2突变的癌症患者受益于ICIs免疫治疗
检索并分析了MSK-IMPACT中接受ICIs(抗PD-1/PD-L1/CTLA-4)治疗的1662例晚期癌症患者的基因组和临床结局数据。476个癌基因中,127个基因与免疫治疗的有益效果相关(p<0.05)。排名靠前的基因包括已知的ICIs治疗预测性生物标志物,包括TP53、PAK7、TET1、PTPRD/PTPRT和STK11。SETD2在476个基因中名列前三。SETD2失活突变的患者在免疫治疗中有更有利的结果(图7A、B),这与先前的报道一致,这一结果通过黑色素瘤(图7C、D)和非小细胞肺癌队列进行了验证。
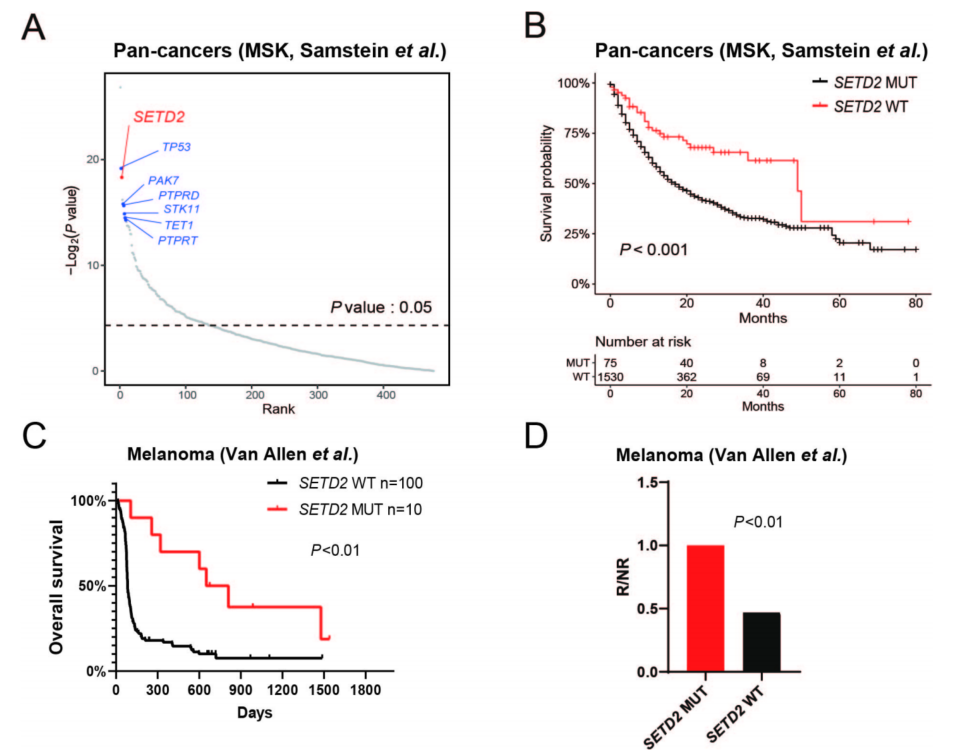
图7 癌症SETD2突变的患者受益于免疫检查点抑制剂(ICIs)免疫治疗
三、研究结论
ETD2失活通过减少NR2F1转录,从而激活STAT1信号通路,促进趋化因子和PD-1的表达并增强抗原提呈,增强免疫效果。携带SETD2突变的癌症患者接受了ICIs,显示出持久的临床获益和生存率增加。
参考文献:
Tumor cell-intrinsic SETD2 inactivation sensitizes cancer cells to immune checkpoint blockade through the NR2F1-STAT1 pathway.[J]Journal for Immunotherapy of Cancer, 2023.