
| 时间:2024-09-06 |
2023年发表于《British Journal of Cancer》(IF=8.8)
一、研究背景
胰腺导管腺癌(Pancreatic ductal adencarcinoma, PDAC)缺乏有效的治疗方法,是一种预后较差的恶性肿瘤。因此,迫切需要进一步研究PDAC的发病机制,并确定额外的治疗靶点以获得更有效的治疗。表观遗传变化,如染色质可及性和组蛋白修饰,在控制转录程序的变化中起着至关重要的作用。表观遗传通路的改变是肿瘤进展的一种新机制。
二、研究结果
1、PDAC小鼠模型中可及染色质的动态变化
采用KC和KPC小鼠研究胰腺癌的表观遗传变化(图1a),对WT、KC和KPC小鼠(每组n=2)进行了ATAC-seq、H3K27ac ChIP-seq和RNA-seq。与WT小鼠相比,KC和KPC小鼠可及性变化趋势类似(图1b),如肿瘤抑制基因Runx3可及性降低(图1e),而致癌基因Egfr可及性升高(图1e)。使用BETA将基因表达变化与染色质可及性信号整合,分别对WT、KC和KPC小鼠进行两两比较。四个基因的表达水平与相应的染色质可及性呈正相关(图1e, f)。
将所有35452个差异可达区域划分为5个聚类。簇2和簇5富集WT,簇3富集KC,簇1和簇4富集KC有利的KPC(图1h)。聚类1、3和4与RNA表达的显著变化呈正相关,KEGG分析表明簇1和簇4有利于KPC,并且在ECM受体相互作用和钙信号通路中富集,但与簇2和簇5相比,在免疫相关信号通路中缺失(图1i)。这些结果表明,染色质可及性的改变可能通过抑制免疫相关信号传导促进PDAC肿瘤的发生和进展。
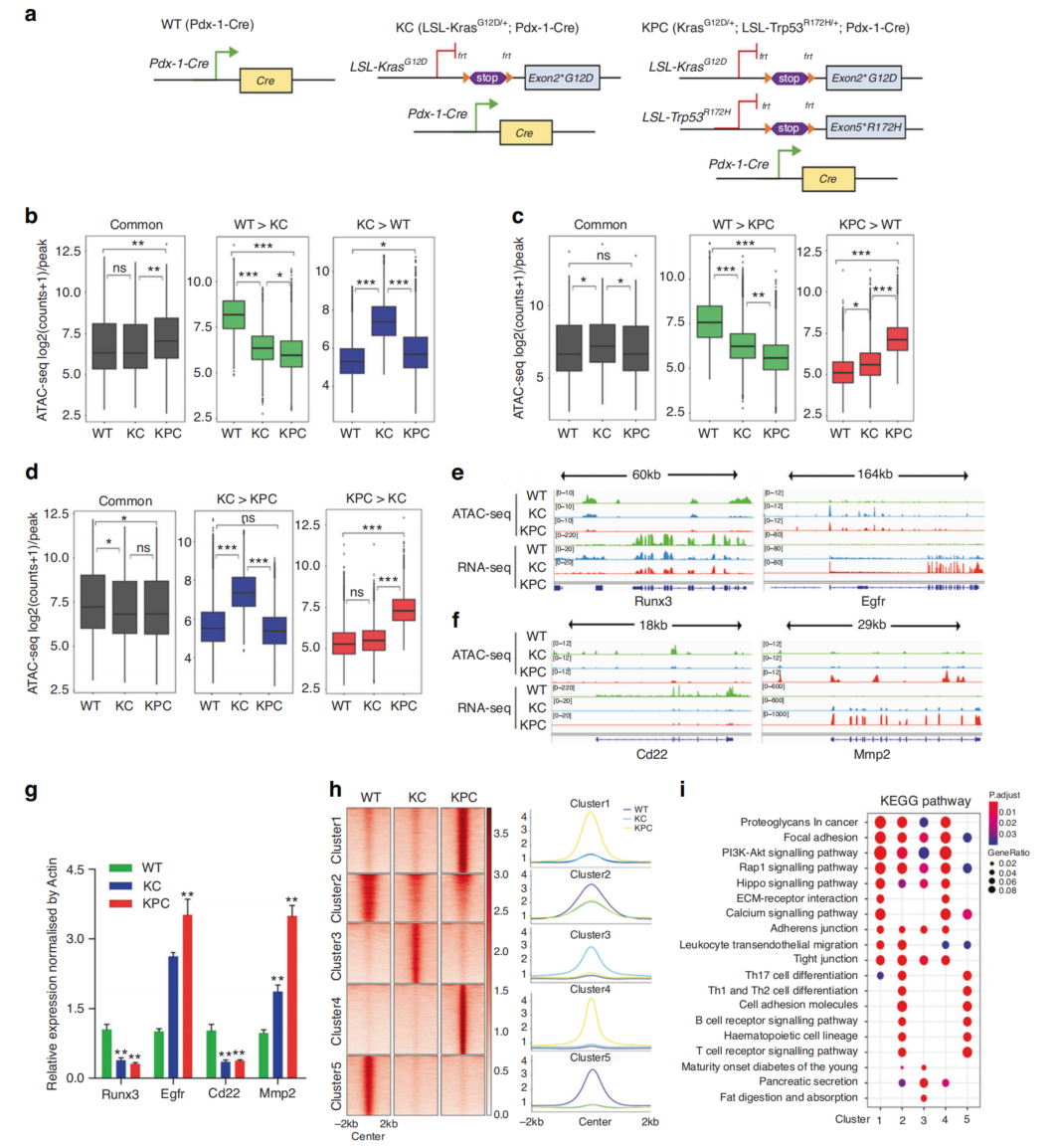
图1 PDAC小鼠模型中可及染色质的动态变化
2、PDAC小鼠的H3K27ac差异谱
H3K27ac峰在启动子的全局信号在三组之间没有差异(图2a),但与WT小鼠相比,KPC和KC小鼠的基因间区域信号逐渐升高(图2b)。KPC与KC中仅鉴定出633个H3K27ac差异峰(图2c),表明KPC与KC小鼠之间H3K27ac信号差异很小。尽管KPC和KC小鼠之间的H3K27ac差异峰数量较少(图2c),但大多数这些峰都是KC特异性的(图2f)。与ATAC-seq和RNA-seq的GO结果一致,免疫相关途径,如T细胞活化、白细胞-细胞粘附和淋巴细胞分化,在KC和KPC小鼠中被抑制或缺失(图2i),这意味着免疫信号的抑制在PDAC的进展中发挥了重要作用。
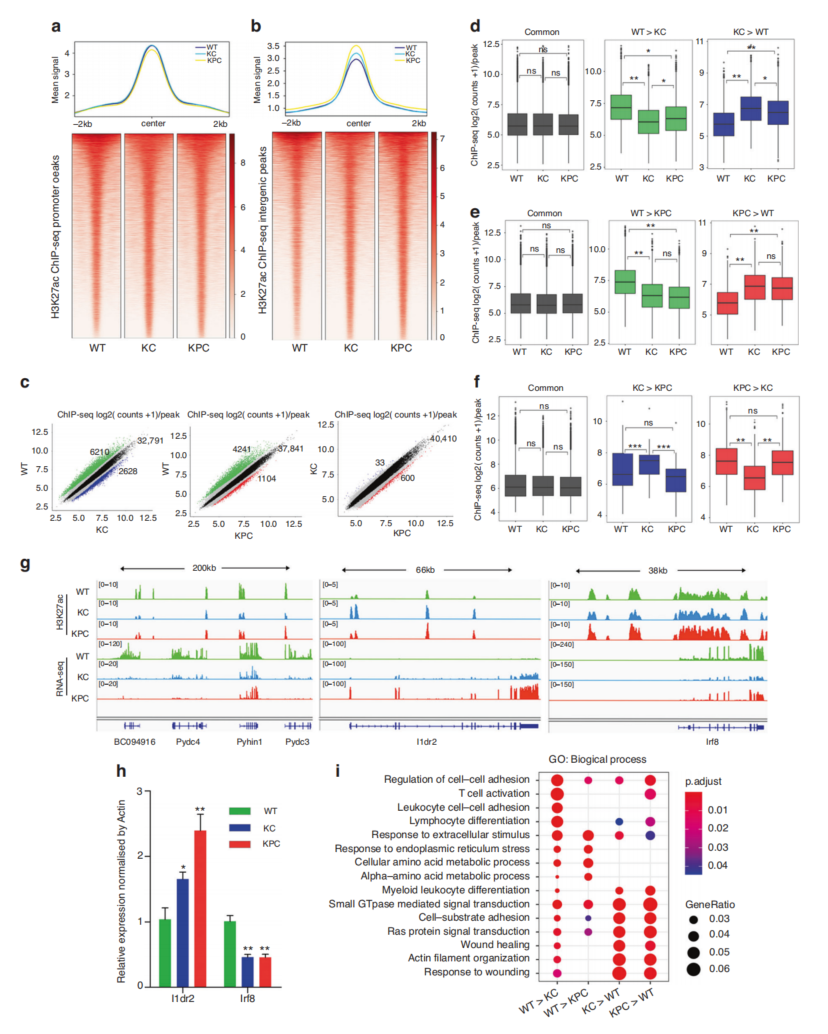
图2 PDAC小鼠模型中H3K27ac信号的变化
3、对差异可及区域的基序分析揭示了新的调节因子
顺式调节元件通常通过与TF结合在肿瘤的发生和进展中发挥重要作用。集群1和4富集了bZIP和TEAD基序,集群2和5富集了IRF和EST家族,集群3富集了Forkhead和Zf TF(图3a)。与WT相比,簇3中富集的大多数TF在KC和KPC中的表达差异更大(图3b-d)。重点研究了KPC中富集的TF,以及KPC和KC中比WT小鼠表达更高的TF(图3e)。与WT相比,Fosl2在KC和KPC中有更高的染色质开放性、H3K27ac信号以及RNA表达(图3f)。通过RT-qPCR、Western blotting和IHC证实,与WT小鼠相比,KPC和KC中FOSL2的表达更高(图3g-i)。因此,将重点放在FOSL2上进行进一步的研究。
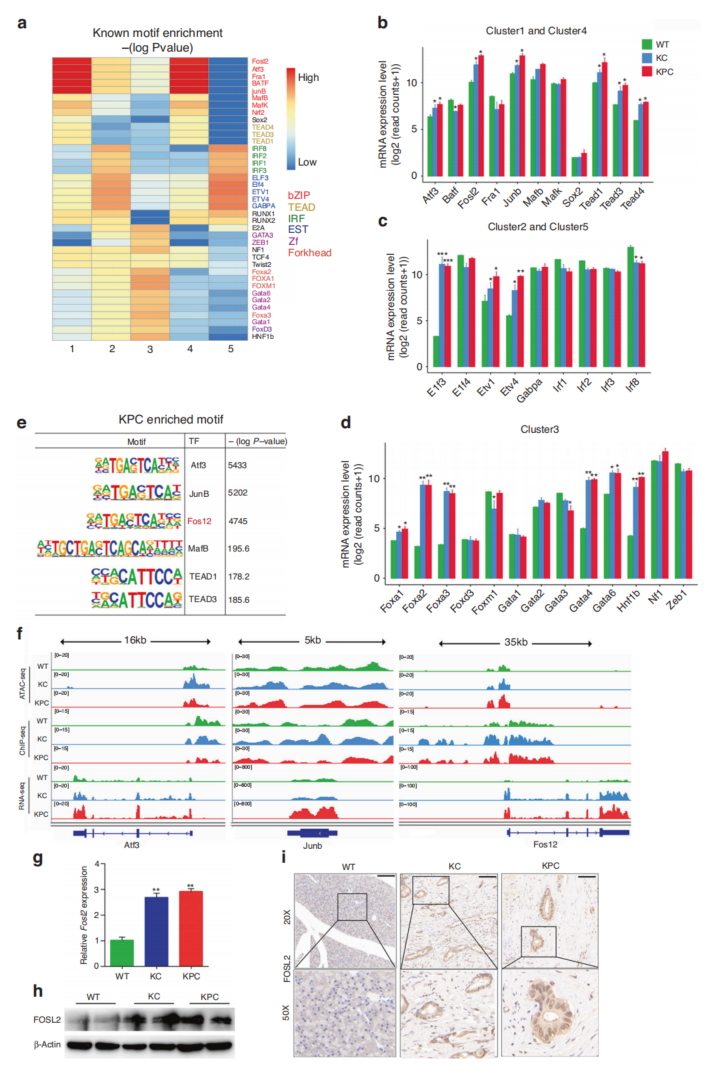
图3 在PDAC小鼠模型中,TF FOSL2的表达上调
4、FOSL2表达上调,并与PDAC患者预后不良相关
使用三个GEO数据集和TCGA的数据,结果显示,与正常样本相比,PDAC样本中FOSL2 mRNA的表达量很高(图4a)。免疫组化分析FOSL2在PDAC组织中的表达明显上调(图4b, c)。此外,FOSL2高表达与预后不良呈正相关(图4d),单因素和多因素Cox比例风险回归分析表明,FOSL2是PDAC总生存期(OS)的独立预后因素。
为了探索FOSL2表达的特定细胞类型,我们重新分析了24个PDAC肿瘤的公开scRNA-seq数据组织和11控制胰腺组织。进行了聚类和细胞类型注释分析(图4f),FOSL2在T细胞、B细胞和腺泡细胞中的表达非常低,但在内皮细胞、导管细胞和其他类型的细胞中表达丰富(图4,h)。肿瘤样本的部分导管细胞中MUC5AC的表达丰富(图4i, j),肿瘤样本的导管细胞中FOSL2的表达上调(图4i, k)。PDAC导管细胞中MUC5AC和FOSL2的表达呈正相关(图4j, k)。这些结果表明,与正常胰腺导管细胞相比,FOSL2在癌性胰腺导管细胞中表达上调,可以作为PDAC患者预后的一种新的生物标志物。
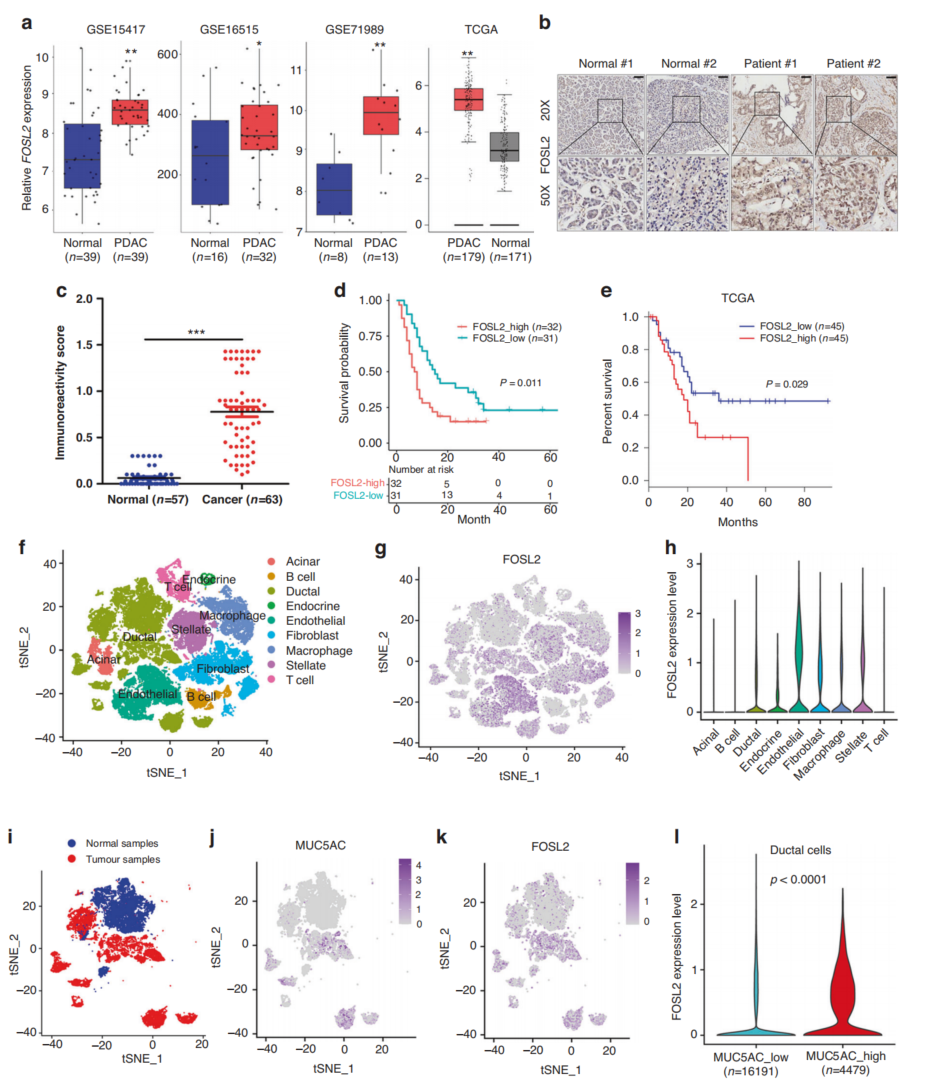
图4 在临床PDAC样本中,FOSL2表达上调,并与不良预后相关
5、FOSL2促进PDAC的进展和免疫逃避
根据FOSL2的表达将scRNA-seq中的导管细胞分为两组。高FOSL2组定义为原始FOSL2计数大于零,其余导管细胞归类为低FOSL2组(图5a)。参与Ras信号转导、上皮细胞迁移和MAP激酶活性的途径被激活(图5c)。还通过实验研究了FOSL2在体内和体外的生物学功能。与对照组相比,FOSL2敲低降低了MIA PaCa-2和PANC-1细胞的细胞增殖。相反,FOSL2过表达促进了MIA PaCa-2、PANC-1和PanO2细胞的细胞增殖。高FOSL2组的免疫信号被抑制(图5d),这意味着FOSL2可能通过抑制肿瘤免疫微环境来促进PDAC的进展。FOSL2过表达显著加速了肿瘤生长(图5f-h),e-FOSL2肿瘤中CD8+ T细胞浸润显著减少(图5i, k)。总之,这些结果表明FOSL2可能驱动PDAC进展和免疫逃避。
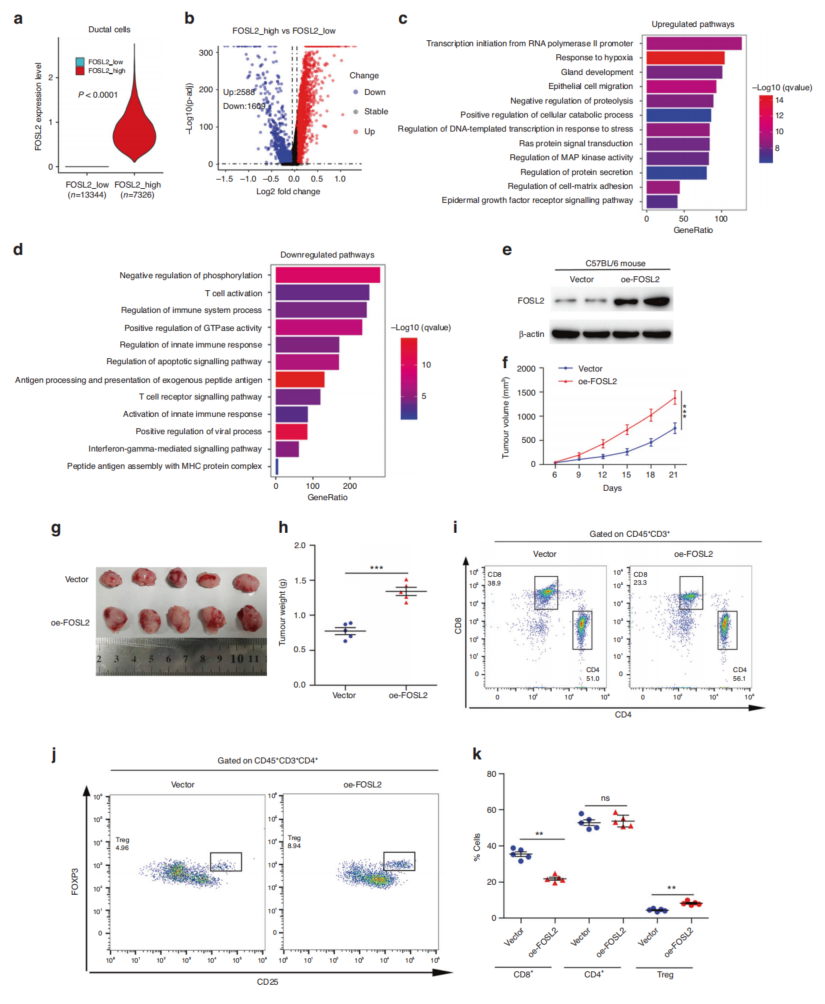
图5 FOSL2可促进PDAC的免疫抑制
6、FOSL2通过与上游的CCL28结合,转录激活CCL28
为研究FOSL2在PDAC中的靶基因,用FOSL2抗体进行了CUT&Tag,以在全基因组范围内确定其结合位点。FOSL2定向调控发生在启动子和增强子上(图6a)。将FOSL2结合区域与KPC转录组中的上调基因相交,CCL28基因在其上游区域的FOSL2占用率高于其他靶点(图6b)。既往报道表明,CCL28不仅参与粘膜免疫,还参与肿瘤免疫抑制。本研究的结果还表明,FOSL2可以调节PDAC中的免疫细胞浸润(图5i-k)。这些发现暗示CCL28介导FOSL2在塑造肿瘤免疫微环境中的作用。因此,探索CCL28是否是FOSL2的真正靶点。
与WT小鼠相比,KC和KPC小鼠的CCL28表达上调(图6e),并与FOSL2呈正相关。CCL28主要在胰腺导管细胞中表达(图6g)。进一步研究了FOSL2上调CCL28的分子机制。选择了四个假定的FOSL2结合位点(-1709 bp,-4192 bp,-8200 bp和-11,414 bp),ChIP-qPCR分析显示,只有结合位点3(BS3)在FOSL2中富集(图6n、o)。在CCL28-8~-12 kb区域构建野生型(WT)和突变型(Mut)CCL28荧光素酶报告质粒,并进行DLR基因检测(图6p)。结果表明CCL28是FOSL2的直接转录靶点,其转录激活是通过FOSL2与BS3结合而发生的。
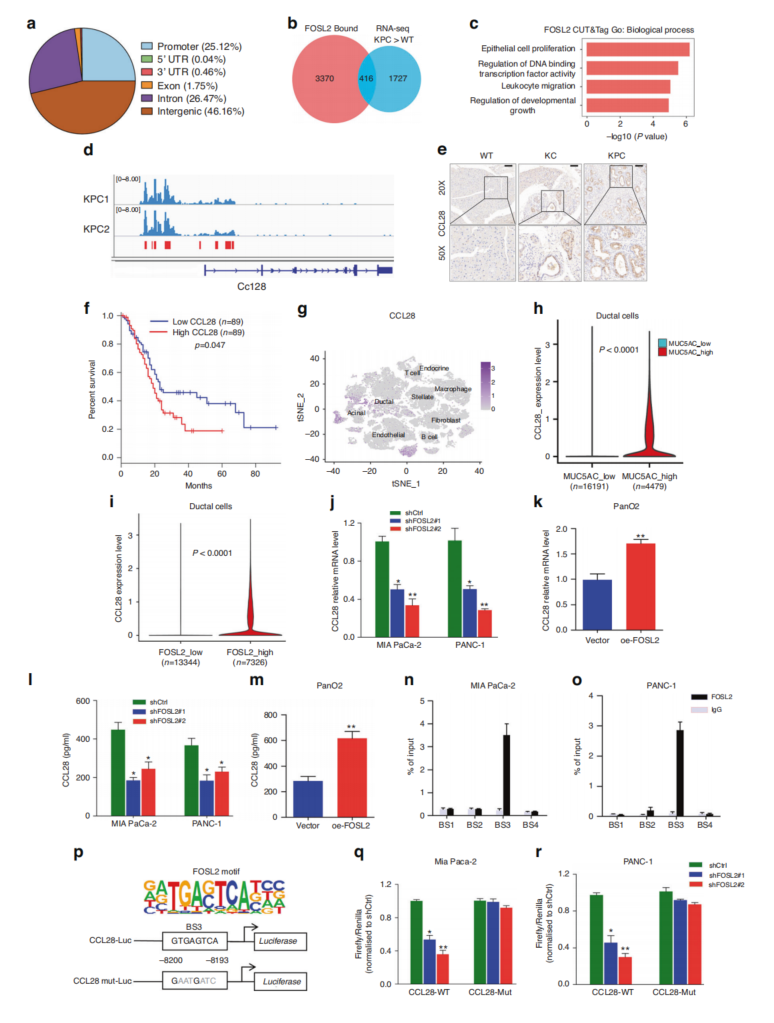
图6 CCL28是FOSL2的一个转录靶点
7、FOSL2通过CCL28促进肿瘤的生长和Treg细胞的募集
有报道称CCL28可募集Treg细胞,引起肿瘤免疫抑制。FOSL2可以在体内招募Treg细胞(图5j)。因此,想探究FOSL2是否通过CCL28在PDAC中募集Treg细胞。与同型对照相比,抗CCL28治疗显著减小了肿瘤大小(图7b-d)。抗CCL28处理小鼠肿瘤组织中Treg细胞比例显著降低(图7e),但抗CCL28处理小鼠肿瘤组织中CD8+和CD4+ T细胞比例未发生变化(图7f, g)。与对照组相比,FOSL2过表达小鼠血清中的血管内皮生长因子a(VEGFA)含量显著增加,但在FOSL2过表达后,通过抗ccl28治疗得以恢复(图7小时)。综上所述,这些结果表明CCL28介导fosl2驱动的肿瘤生长和Treg细胞浸润。
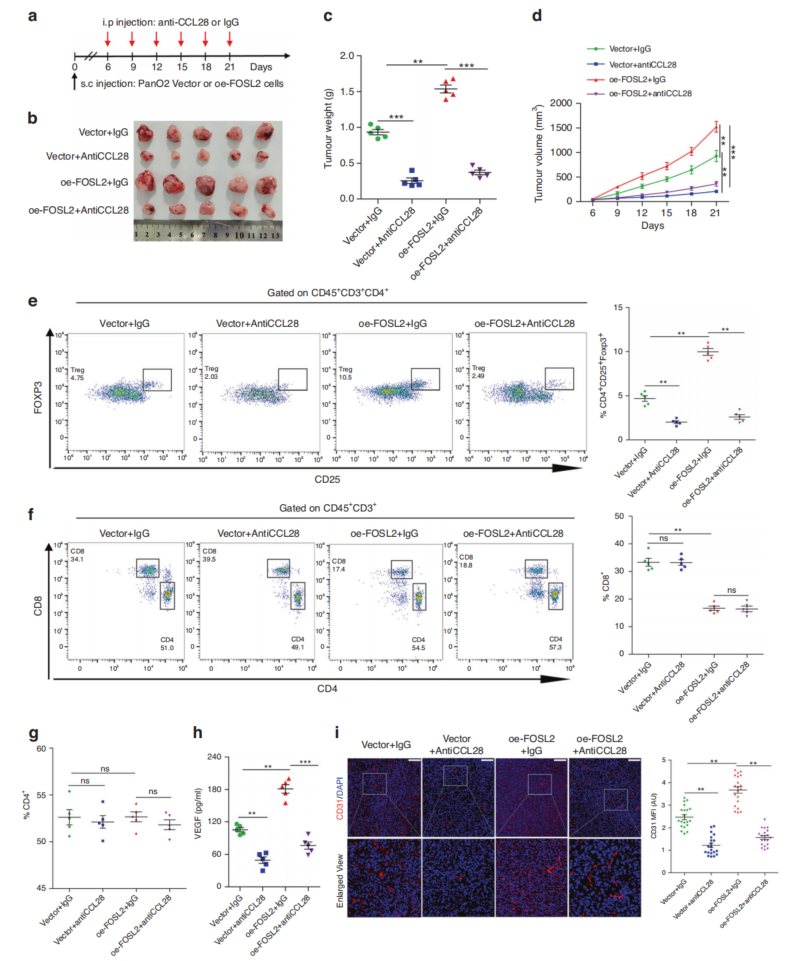
图7 FOSL2通过CCL28促进Treg细胞的募集
8、突变体KRAS通过MAPK/ERK信号通路调控FOSL2的表达
根据scRNAseq数据,KRAS在内皮细胞和导管细胞中的表达高于其他类型的细胞。为了进一步研究KRAS与FOSL2之间的关系,进行了KRAS敲除。KRAS缺失导致FOSL2 mRNA和蛋白水平下调(图8a, b),过表达增加了FOSL2 mRNA和蛋白水平(图8a, c)。scRNA-seq显示,在PDAC导管细胞中,FOSL2与KRAS呈正相关。KRAS的缺失降低了CCL28的mRNA和蛋白水平表达(图8a、d、f)。这些结果表明,KRAS调节FOSL2及其靶点CCL28的表达。已知RAS激活可驱动RAF/MEK/ERK级联反应,为了了解KRAS上调FOSL2的机制,用了MEK(Selumetinib)和ERK(SCH772984)的特异性抑制剂。使用MEK抑制剂处理后,FOSL2的表达降低(图8h, k)。这些发现表明突变体KRAS通过MAPK/ERK途径介导FOSL2转录和随后的CCL28表达。
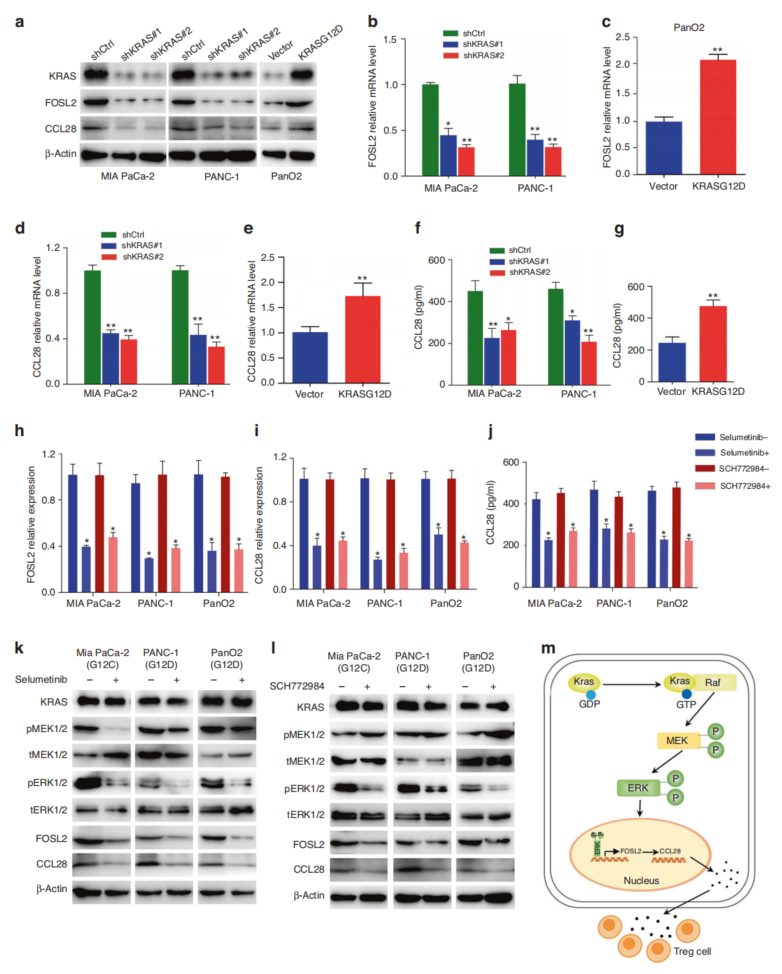
图8 KRAS通过MAPK/ERK通路上调FOSL2的表达
三、研究结论
KRAS驱动的FOSL2通过转录激活CCL28促进PDAC的进展,揭示了FOSL2在PDAC中的免疫抑制作用。
参考文献:
Chromatin accessibility uncovers KRAS-driven FOSL2 promoting pancreatic ductal adenocarcinoma progression through up-regulation of CCL28.[J]British Journal of Cancer, 2023.