
| 时间:2024-06-27 |
2023年发表于《Theranostics》(IF=12.4)
一、研究背景
急性肾损伤(AKI)是一种多因素介导的肾脏疾病,脓毒症所致AKI的主要病理过程是肾小管上皮细胞(TECs)的凋亡和肾间质炎症。很多研究表明,表观遗传调控在AKI和损伤修复中起着关键作用,其中组蛋白甲基化修饰可以上调或下调基因转录。组蛋白H3K27被EZH2甲基化,导致靶基因的转录沉默。有证据表明EZH2介导的组蛋白修饰有助于AKI。然而,目前EZH2在脓毒症AKI中的潜在机制和确切功能尚不清楚。Wnt/β-catenin信号通路是一种进化高度保守的信号通路,已有研究证实了Wnt/β-catenin通路在多种肾脏疾病中具有多种致病功能。本研究发现沉默EZH2后,可通过减缓Sox9的转录抑制、激活Wnt/β-catenin通路、减轻肾间质小管上皮细胞凋亡和炎症反应,从而保护肾功能;靶向EZH2对治疗脓毒症诱导的AKI有重要的临床价值。
二、研究结果
1、脂多糖(LPS)处理小鼠肾脏诱导EZH2
实验组小鼠腹腔注射LPS(15mg/kg)。24h后,western blotting显示凋亡相关蛋白caspase3和Bax表达水平被激活,抗凋亡蛋白Bcl2表达水平下降(图1A)。实验组小鼠血清肌酐(SCr)和血尿素氮(BUN)水平明显升高(图1B),TUNEL荧光显示脓毒症诱导的AKI体内模型成功建立(图1C);模型组TNF-α、IL-6、IL-1β显著上调(图1D)。单核RNA-seq(SNRNA-seq)显示,AKI期间近端肾小管上皮细胞核中EZH2表达上调(图1E)。IF显示EZH2位于近端小管上皮细胞中,与对照组相比,LPS处理组EZH2的表达显著上调(图1F);EZH2表达与SCr水平呈正相关(图1H)。与对照组相比,LPS处理组H3K27me3的表达显著上调(图1G)。

图1 EZH2在脓毒症诱导的急性肾损伤(AKI)体内表达上调
2、敲低EZH2可减轻脓毒症引起的小鼠肾损伤、细胞凋亡和炎症反应
为了进一步探讨EZH2在脓毒症诱导AKI中的生物学机制,使用EZH2iKO小鼠,并通过免疫组化和western blotting检测敲除效率(图2A-B)。LPS会引起许多组织病理学改变(图2C)。与EZH2fl/fl小鼠相比,EZH2iKO小鼠在LPS注射后表现出更小的肾脏损伤;TUNEL组织化学显示EZH2iKO小鼠肾组织中凋亡细胞明显少于EZH2fl/fl小鼠。与对照组相比,LPS引起AKI模型血清BUN和SCr水平显著升高,EZH2iKO小鼠肾功能有所改善,BUN和SCr表达水平降低(图2D)。
与EZH2fl/fl小鼠相比,EZH2iKO小鼠中cleaved-caspase3和Bax的表达下调,而Bcl2的表达上调(图2E),TNF-α、IL-6、IL-1β和MCP-1水平显著降低(图2F)。与EZH2fl/fl小鼠相比,EZH2iKO小鼠的巨噬细胞浸润明显减少,这与炎症因子的变化趋势一致(图2C);CD11bhi/F4/80lo群体减少(图2G)。综上所述,EZH2的下调可能通过减少肾间质巨噬细胞的积聚而发挥抗炎作用。
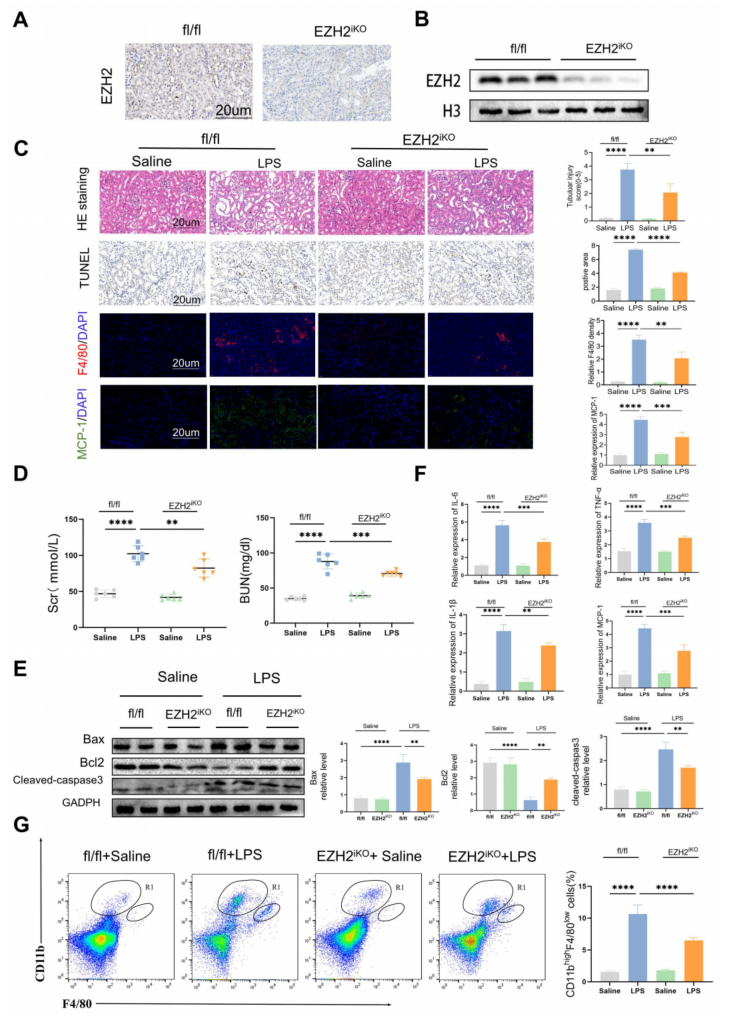
图2 敲除EZH2可减少体外细胞凋亡和炎症反应
3、EZH2敲低可减少LPS诱导的体外细胞凋亡和体外炎症反应
使用sh-EZH2细胞进一步探讨EZH2在脓毒症诱导的AKI中的作用机制,Western blotting验证了EZH2的敲除效率,EZH2 shRNA对细胞凋亡和炎症指标均无影响(图3A-B)。用50µg/mL的LPS处理,细胞活力严重降低;当EZH2被敲除时,LPS处理下HK2细胞活力恢复(图3C)。与体内实验一致,western blot结果显示,cleaved-caspase3和Bax表达受到抑制,Bcl2表达上调(图3D-E),流式细胞也得到了相同的结果(图3F)。这些结果表明,抑制EZH2可减少脓毒症诱导的aki诱导的上皮细胞凋亡。
RT-qPCR结果显示,LPS处理后HK2细胞IL-6、IL-1β和TNF-α mRNA水平显著升高,而sh-EZH2细胞炎症因子表达显著下调(图3G)。在对照组中,HK2细胞几乎没有MCP-1的表达;而LPS处理后,MCP-1大量表达。EZH2敲低后,MCP-1的升高发生逆转(图3H-I)。用LPS刺激是巨噬细胞由上向下迁移的数量明显增加,LPS处理的sh-EZH2中巨噬细胞的迁移数量明显减少了(图3K)。这说明EZH2敲除可以减轻炎症反应。

图3 敲除EZH2可减少体外细胞凋亡和炎症反应
4、EZH2通过表观遗传修饰调控Sox9的表达
取两个重复样本的ChIP-seq取交集(图4A)。Motif分析显示EZH2与富含GC的序列结合较多,说明EZH2可以结合到基因的启动子区域,从而影响基因的转录(图4C)。对EZH2(IP2样本)的ChIP-seq数据和EZH2沉默后HK2细胞中上调基因进行了取交集分析(图4D),GO和KEGG富集分析显示与经典Wnt信号通路密切相关(图4E-F),并且EZH2与Sox9结合(图4G)。之后的ChIP-qPCR验证,发现EZH2富集于Sox9的启动子区域(图4H);EZH2敲低后Sox9的表达更高(图4I)。因此,EZH2可以通过表观遗传学调控Sox9的表达,从而影响脓毒症诱导的AKI。
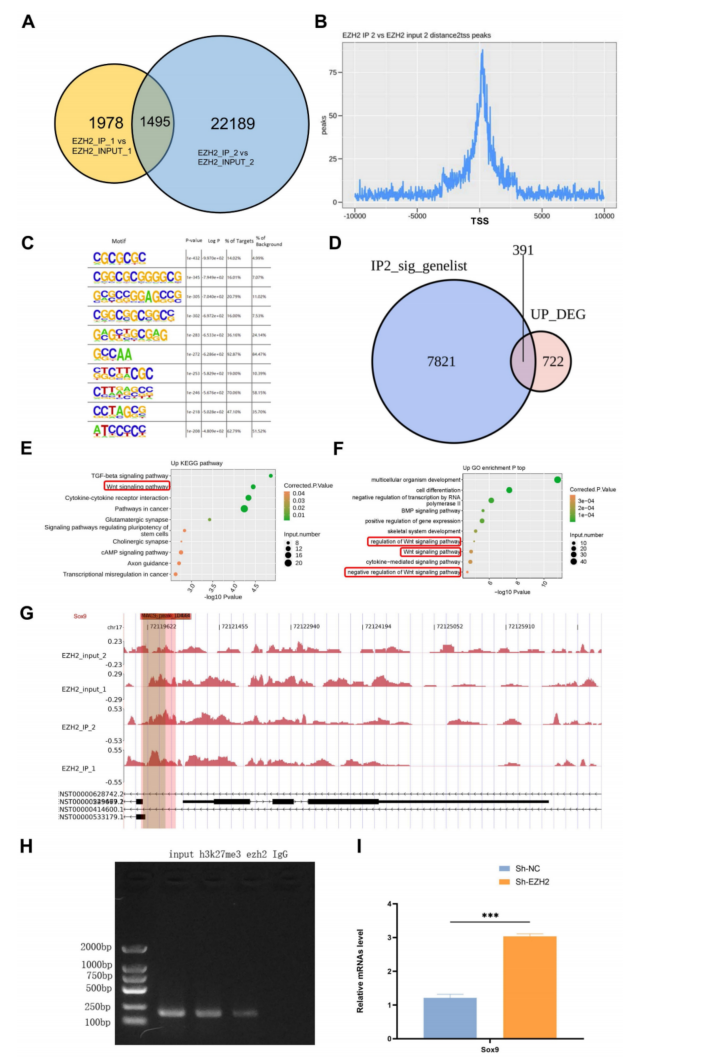
图4 EZH2通过表观遗传效应影响Sox9的转录
5、EZH2通过上调Sox9减轻脓毒症诱导AKI的细胞凋亡和炎症
Sox9敲低(siSox9)导致Wnt/β-catenin通路下游基因survivin的表达下调。Bax的下调被逆转,抗凋亡蛋白Bcl2的下调也被逆转。MCP-1等炎性因子表达明显升高,巨噬细胞迁移能力逆转。使用ICG-001(一种Wnt/β-catenin通路抑制剂),凋亡相关蛋白和巨噬细胞迁移也被逆转(图5D-E)。EZH2被敲除后,Sox9的表达得以恢复,Wnt/β-catenin通路的表达被激活,有助于减轻肾损伤。Transwell实验也表明,敲除Sox9和添加Wnt/β-catenin途径抑制剂后,巨噬细胞迁移发生逆转。EZH2直接作用于Sox9调控Wnt/β-catenin通路,从而影响脓毒症诱导AKI的凋亡和炎症反应。
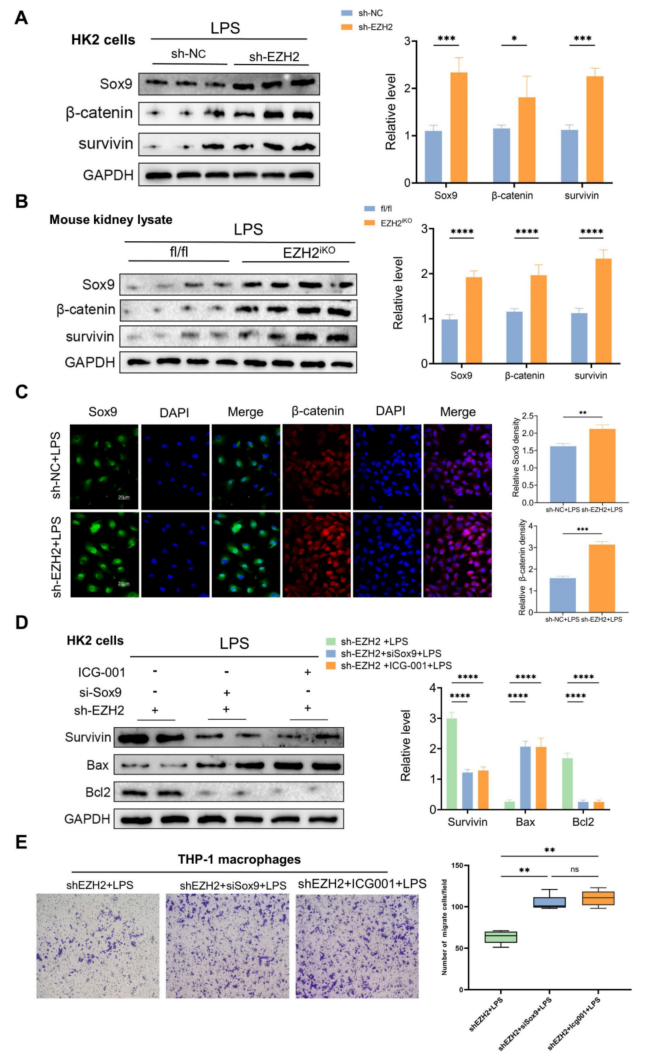
图5 敲除EZH2增加了Sox9及其相关下游通路的表达
6、Sox9通过Wnt/β-catenin通路调控细胞凋亡和炎症反应
通过过表达(Sox9ov)和敲低Sox9来探索β-catenin及其下游表型的变化。发现当Sox9过表达时,β-catenin的表达增加,发生核易位。当Sox9被敲除时,β-catenin的表达降低(图6A-C)。Sox9的过表达也增加了Wnt/β-catenin通路下游基因survivin和CyclinD1的表达,ICG001抑制了这两个基因的表达(图6D)。而Sox9过表达降低了凋亡因子和炎症因子的表达,抑制了巨噬细胞的募集;Wnt/β-catenin通路抑制剂ICG001抵消了Sox9过表达的影响(图6E-G)。
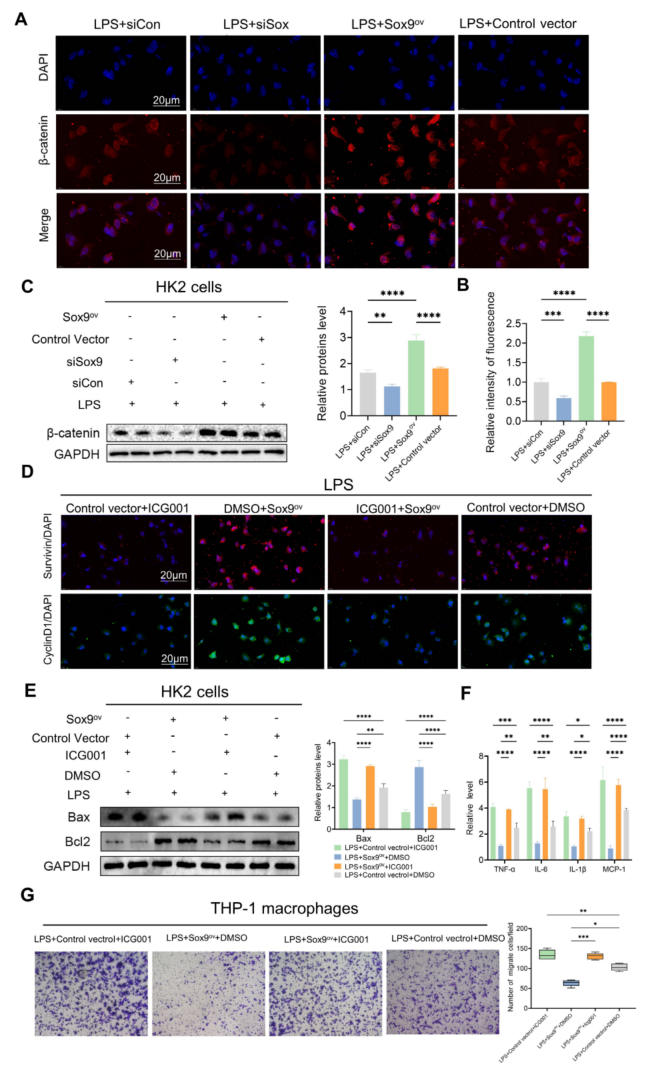
图6 Sox9通过Wnt/β-catenin通路调节细胞凋亡和炎症反应
7、在脓毒症诱导的AKI中,EZH2抑制剂3-DZNep可保护肾功能
使用EZH2抑制剂3-DZNep验证AKI期间EZH2在体内的功能(图7A),LPS+3-DZNep组SCr和BUN水平及肾系数降低,病理肾损害减轻(图7B-D)。TUNEL染色显示3-DZNep可减少肾细胞凋亡。这与Western blotting一致,3-DZNep抑制EZH2、Bax表达,促进Bcl2表达(图7E)。3-DZNep持续下调MCP-1、IL-6、IL-1β和TNF-α水平(图7F)。IF显示EZH2抑制剂可减少巨噬细胞的浸润(图7G)。采用CLP构建脓毒症AKI模型也得到了一致的结果。这些数据表明3-DZNep可以预防AKI并减少炎症反应。
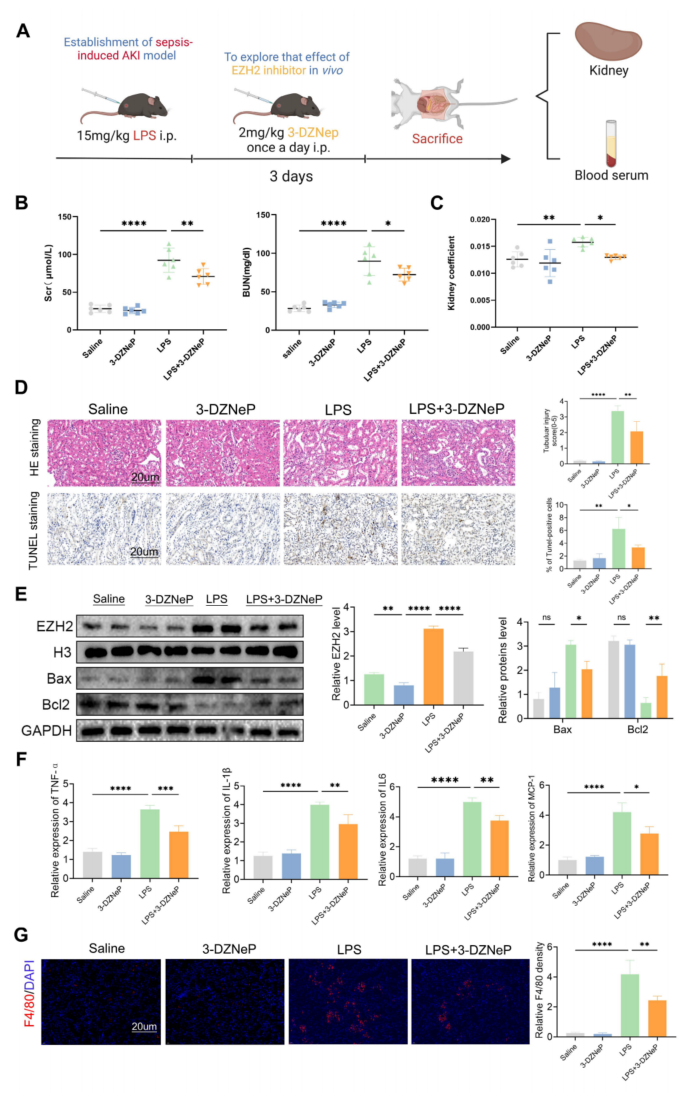
图7 EZH2抑制剂3-DZNep通过抑制细胞凋亡和炎症反应来保护脓毒症诱导的AKI的肾功能
三、研究结论
本研究证实沉默EZH2后,可通过减缓Sox9的转录抑制、激活Wnt/β-catenin通路、减轻肾间质小管上皮细胞凋亡和炎症反应,从而保护肾功能;靶向EZH2对治疗脓毒症诱导的AKI有重要的临床价值。
参考文献:
Histone H3K27 methyltransferase EZH2 regulates apoptotic and inflammatory responses in sepsis-induced AKI.[J]Theranostics, 2023.