
| 时间:2024-06-14 |
2023年发表于《Nature Communications》(IF =16.6)
一、研究背景
肾细胞癌(RCCs)是最常见的肾肿瘤类型,可分为多个组织学亚型:透明细胞RCC (ccRCC)约占75%,乳头状RCC (pRCC)和嫌色RCC (chRCC)分别占约15%和5%。ccRCC、pRCC和chRCC的基因组和转录组学研究,揭示了RCC不同亚型之间显著的异质性,每种亚型都表现出不同的体细胞突变、染色体拷贝数改变和基因表达谱。组蛋白修饰和染色质可及性与不同RCC组织亚型的关系,以及与肾癌遗传性的关系,尚未有系统研究。
增强子是顺式作用的DNA区域,翻译后组蛋白修饰(如H3K27ac和H3K4me2)的ChIP-seq已经在哺乳动物基因组中鉴定出数百万个增强子。分析增强子已成为表征驱动细胞转录状态的关键转录因子(TF)和更好地理解种系风险变异的有力工具。细胞的身份和功能主要由主转录组因子决定,主转录因子(MTF)与增强子结合并调节建立细胞身份和功能所需的基因。此外,基于人群的表观基因组分析发现,表达量性状位点(eQTLs)的单核苷酸多态性(SNPs)通常也与附近表观基因组特征(如以H3K27ac标记的活性增强子)的变异相关联。许多先前的RCC表观遗传学数据是基于细胞系的,这些细胞系与原始肿瘤不同,并不代表所有的组织学亚型。
本研究中综合分析了H3K27ac、H3K4me2的修饰数据(ChIP-seq)、染色质可及性分析(ATAC-seq)、DNA-seq和RNA-seq的数据,定义了42例原发性人类RCC肿瘤中不同组织学亚型的RCC的表观遗传图谱和调控网络;分析了不同组织亚型中的特异增强子,亚型特异型的主转录因子(MTF)。
二、研究结果
1、绘制肾细胞癌的染色质调控网络
以研究它们的染色质状态、转录因子结合模式以及基因表达情况,42个(24个ccRCC,6个pRCC,12个chRCC)新鲜冷冻的不同RCC亚型的肿瘤样本进行了ChIP-seq、ATAC-seq、RNA-seq测序分析。其中将患者分为2组(1和2),H3K4me2 ChIP-seq来绘制活性和静止增强子图谱,使用H3K27ac ChIP-seq来绘制活性启动子和增强子图谱。队列1(12 chRCC,6 pRCC,12 ccRCC)所有样本中共鉴定出153,321个启动子-远端(增强子)H3K27ac ChIP-Seq峰,其中大多数(n=136,469)在两种及以上以上组织亚型中共有(图1A,B)。例如,PAX8位点在所有样本中都被H3K27ac标记。PAX8是参与RCC早期肾脏胚胎发生和肿瘤发生的TF,可作为区分RCC与其他恶性肿瘤的临床诊断工具(图1C)。H3K27ac峰的分层聚类(图1D)和PCA分析清楚地区分了RCC的三种组织学亚型,chRCC的H3K27ac图谱与pRCC或ccRCC有明显差异。总共有12908个位点上调:8939个位点是chRCC特异性的,3653个位点是pRCC特异性的,316个位点是ccRCC特异性的(图1E)。这些组织特异性的H3K27ac峰被H3K4me2 ChIP-seq差异标记,并且与开放染色质相关,表明它们是组织特异性的活性增强子。此外,差异表观遗传位点与最近基因表达差异相关(图1F-H)。
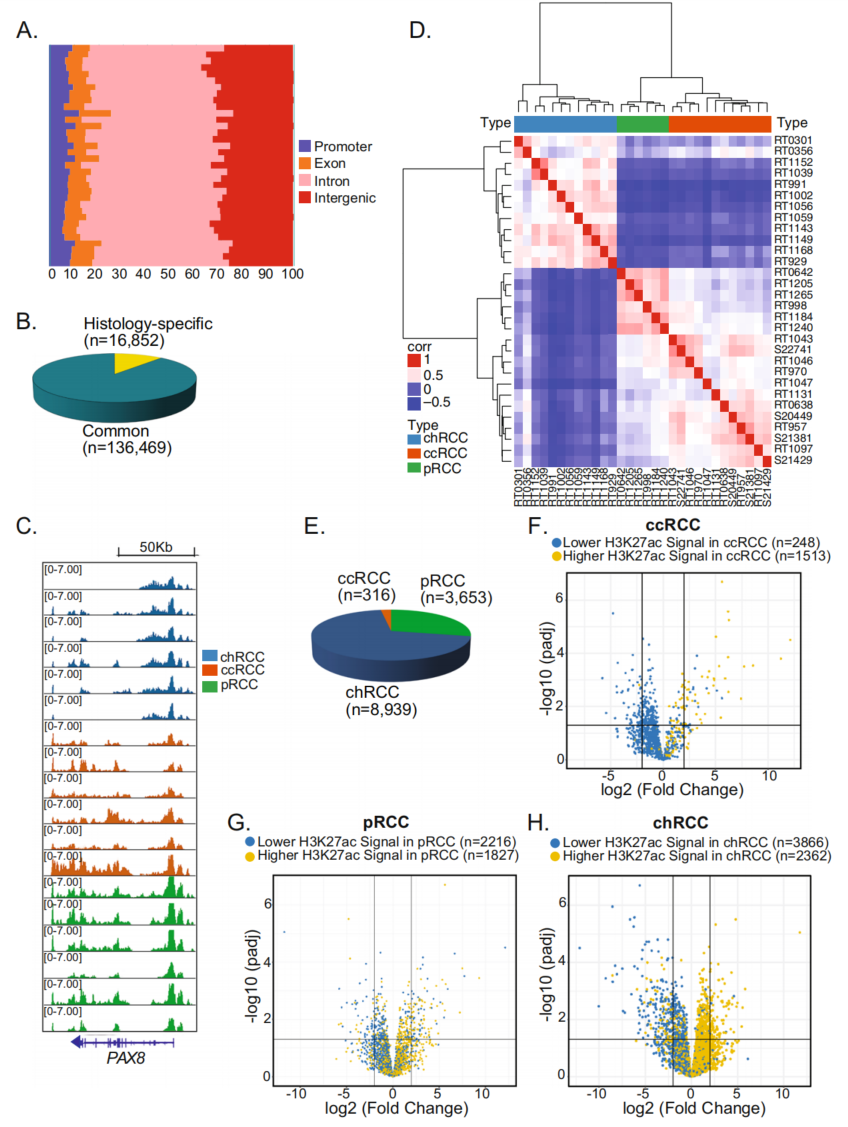
图1 不同组织亚型RCC的H3K27ac图谱
对8939个chRCC特异性H3K27ac峰对应基因的GREAT分析显示,在肌动蛋白调节、脂肪酸氧化和离子跨膜转运蛋白活性中富集(图2A),与先前报道的chRCC中离子跨膜转运特征增加的mRNA特征分析一致。对3653个pRCC特异性位点对应基因富集于肾脏系统发育有关(图2B)。由于与chRCC和pRCC相比,ccRCC的特异性位点相对较少(n=316),并且ccRCC的增强子景观与pRCC更相似,因此我们仅比较了ccRCC和pRCC之间的H3K27ac位点。1265个ccRCC特异性峰与参与循环系统发育和血管生成的基因相关(图2C);而2661个pRCC特异性峰与参与肾脏胚胎发生的基因相关。所有样本的H3K27ac和H3K4me2 ChIP-seq信号呈强相关(图2D)。
对每个亚型中富集的H3K27ac峰进行motif分析,发现了四个在chRCC中高度富集的motif,其中一个类似于叉头(forkhead)基序,另一个类似于与TFCP2相关的基序(图2E)。FOXI1是forkhead TF家族的成员,TFCP2L1与TFCP2密切相关,它们都与肾的插层细胞发育有关,插层细胞是chRCC的起源细胞。在chRCC中H3K27ac信号在FOXI1和TFCP2L1基因位点显著富集,而在ccRCC和pRCC中无信号或信号明显较低(图2F,G)。对ccRCC特异性增强子的motif富集分析发现,一个基序类似于基本亮氨酸拉链(bZIP)BATF基序,另一个基序类似于基本螺旋-环-螺旋(bHLH)TF家族成员HIF2α(也称EPAS1)(图2H)。具有BATF的bZIP家族中的ETS1与von-Hippel Lindau(VHL)依赖性ccRCC的肿瘤发生有关,并且在ccRCC中被H3K27ac高度富集,在pRCC中较少,而在chRCC中则没有(图2I)。众所周知,EPAS1在ccRCC中由于VHL蛋白功能的丧失而失调。它是ccRCC的主要驱动因素,是多个关键致癌途径的上游。最近的临床试验显示HIF2抑制剂在晚期ccRCC患者中具有临床效用。得分最高的pRCC特异性motif对应于HNF1β(图2J,K)。HNF1β是含有同源结构域的TF超家族的成员,参与肾脏发生,并且在pRCC中被H3K27ac高度富集(图2K)。
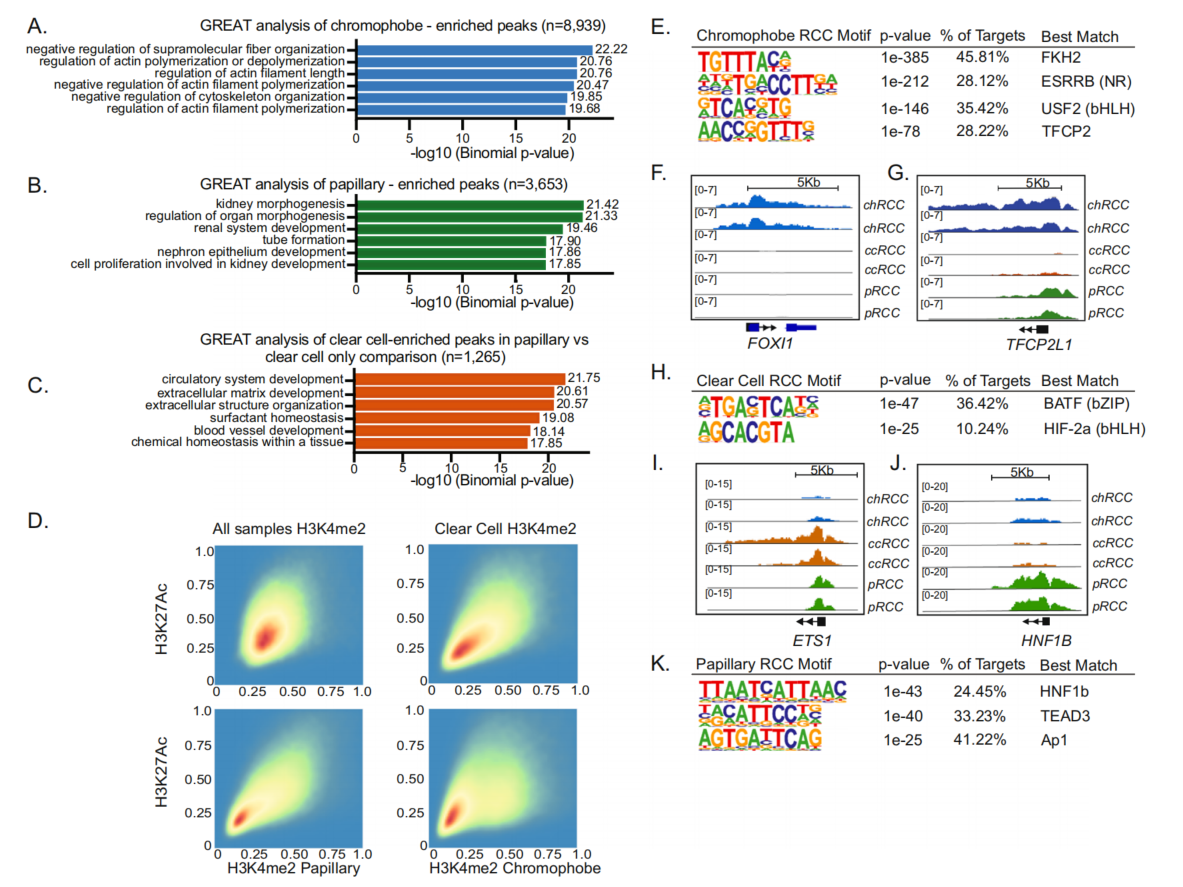
图2 调控元件的表观遗传学注释识别了组织学特异性通路和TFs的富集
2、特定的主转录因子(MTF)定义了RCC的亚型
接下来系统研究三种RCC组织学亚型潜在的特异性MTF。MTF通常结合在SEs中,由SEs调控,并在转录核心调控回路(CRC)中相互调控。本研究利用RNA-seq、ChIP-seq和ATAC-seq数据来鉴定潜在的组织学特异性MTF(图3A)。结合了(1)不同RCC组织学亚型差异表达TF的表达数据;(2)相对于其他癌症类型,RCC组织学亚型特异性TF(CaCTS);(3)不同RCC组织学亚型间差异SE相关TF;(4)在调控网络中具有组织学特异性的TF。这四项分析确定了200多个候选TF,对下游验证进行了优先排序选择了50个候选组织学特异性MTF(N=20,chRCC;N=14,pRCC;N=16,ccRCC),包括FOXI1、TFCP2L1和DMRT2(chRCC);EPAS1、ETS1、BARX2、ZNF395(ccRCC),HNF1B和NR2F2(pRCC)(图3B)。TCGA KICH、KIRC和KIRP队列的基因表达数据集验证了正常组织中没有该TF特异性。为了在蛋白质水平上验证概念,通过免疫组织化学(IHC)研究了MTF的表达特异性和定位。选择了四个具有代表性的MTF(BHLHE41、HNF1β、NKX6.1和ZNF395),发现不同组织学亚型的蛋白表达水平有显著变化(图3C)。更具体地说,两个ccRCC特异性MTF(BHLHE41和ZNF395)在ccRCC肿瘤中高度表达。接下来,通过检查ccRCC和chRCC中的EPAS1细胞,确认了MTF EPAS1在体内的组织学特异性结合(图3D)。亚型特异性EPAS1结合位点高度富集于H3K27ac亚型特异性位点。ccRCC特异性EPAS1结合位点富集于免疫细胞和白细胞活化途径,而chRCC特异性EPAS1结合位点富集于代谢过程和脂肪酸活化途径(图3E,F)。
为了进一步验证本研究的方法,即特定的MTF驱动不同RCC组织学的转录特性,在ccRCC细胞系中操纵TF表达。假设过表达ccRCC特异性TF和抑制ccRCC特异性TF可以改变ccRCC细胞系786-O的转录图谱,使其变得更像chRCC。在MTF分析中,FOXI1评分为chRCC特异性TF(图3B),EPAS1对ccRCC具有高度特异性(图3B),并且先前的研究已经说明了它在ccRCC51发病机制中的作用。对786-O中FOXI1 OE/EPAS1 KD下调基因的基因集富集分析(GSEA)显示,与免疫应答相关的基因富集(图4A)。在786-O细胞中,伴随的FOXI1 OE和EPAS1 KD导致chRCC特异性主转录因子(MTF)如TFCP2L1和NR3C2的上调,以及ccRCC特异性主转录因子(MTF)如ETS1和ZNF395的下调(图4B)。进一步的监督分析表明,与EPAS1 KO/FOXI1 OE细胞株相比,EPAS1的下游靶点CAIX在WT 786-O细胞株中显著过表达。相比之下,与WT 786-O细胞系相比,CD117在EPAS1 KO/FOXI1 OE细胞系中过表达(图4C)。
然后,比较了实验条件下786-O和本研究RCC肿瘤表达数据之间的差异表达基因(图4D)。将chRCC和ccRCC基因集定义为每种组织学中相对于其他组织的前100个上调基因,我们发现与对照相比,786-O FOXI1 OE/EPAS1 KD细胞系中chRCC基因集在上调基因中富集(图4E),而786-O对照与FOXI1 OE/EPAS1 KD细胞系中ccRCC基因集在上调基因中富集(图4F)。FOXI1 OE/EPAS1 KD 786-O细胞系显示,chRCC特异性候选MTF:TFCP2L1、GATA2、DDIT3、NKX6-1的表达显著升高,ccRCC特异性候选MTF:ZNF395和TSC22D3的表达显著降低。总之,这些数据表明,在ccRCC细胞系786-O中,无论是否敲低EPAS1,单一chRCC候选主转录因子(MTF)FOXI1的过表达都会导致显著的表达变化,从而使该细胞系更像chRCC细胞系。
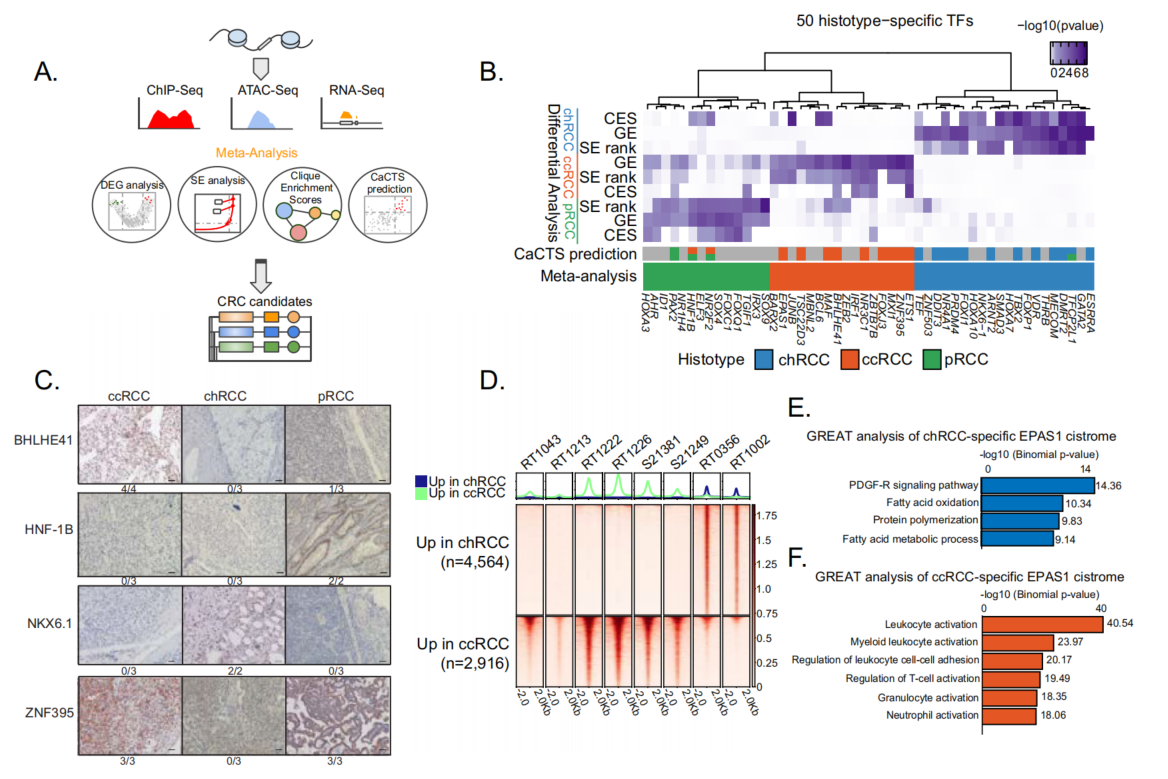
图3 多维整合分析识别了组织学特异性的主转录因子(MTF)
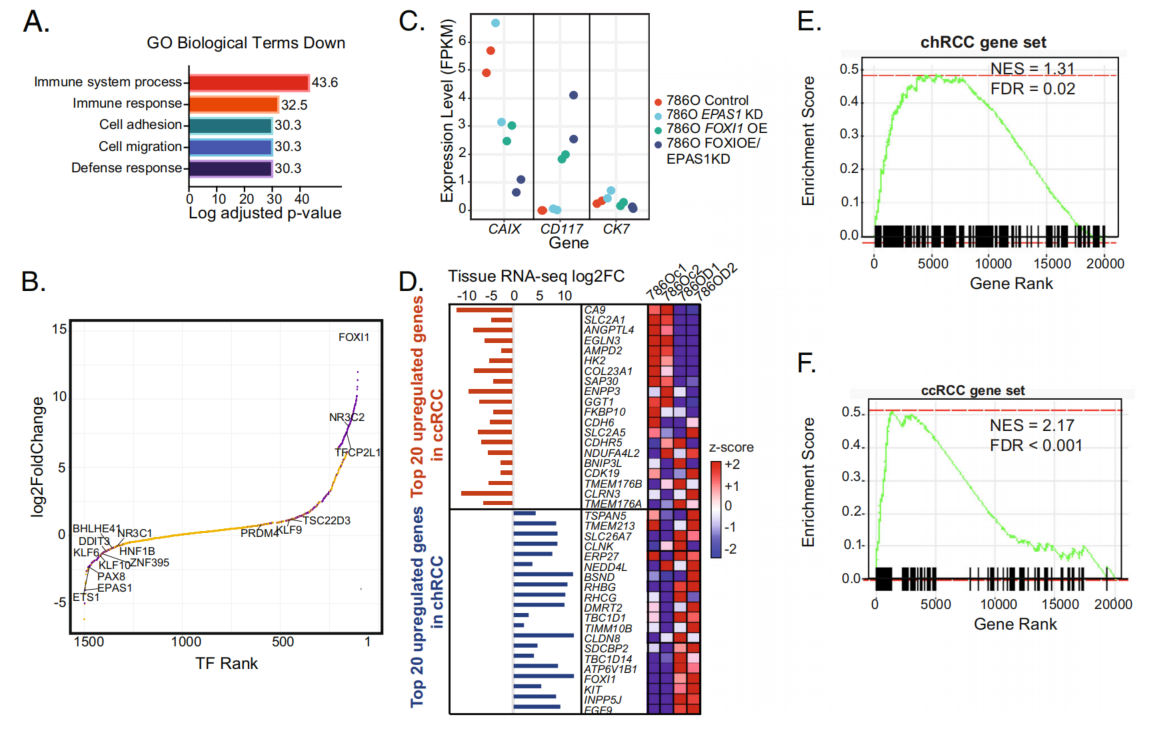
图4 在一个ccRCC细胞系中,两个主转录因子(MTF)的表达变化会导致细胞系更类似于chRCC的转录谱
3、等位基因失衡反应了遗传性肾细胞癌的风险变异
等位基因失衡,即ChIP-seq reads中杂合子差异等位基因的单核苷酸多态性(SNPs),提供了两种单倍型之间顺式调控活性的体内比较(图5A)。因此,等位基因失衡可以突出通过GWAS候选因果变异与RCC风险相关的位点上的功能相关性。分析许多个体表观基因组的一个关键优势是能够捕获杂合位点,并通过分析等位基因失衡来测量TF结合对调控元件的影响。为此,本研究评估了来自这些RCC样本的ChIP-seq数据中的染色质等位基因失衡,以便从RCC的GWAS中发现因果风险SNPs。将stratAS32应用于20个ccRCC和6个pRCC的H3K27ac ChIP-seq数据,在ccRCC和pRCC183099个峰中,确定了10605个染色质不平衡的H3K27ac峰,将其定义为多个假设检验校正后具有一个或多个不平衡SNPs的峰(图5B)。
假设染色质AI峰与主转录因子(MTF)结合的调控元件相对应。在这些元件中,反式作用因子(即MTF)的存在可能导致调控元件活性,可观察到H3K27ac AI峰值。例如rs4903064(chr14:73279420;14q24.2),DPF3的eQTL(图5C)。rs4903064在我们的H3K27Ac数据集中是等位基因不平衡的,已被证明在所有三种组织学亚型(ccRCC,chRCC和pRCC)中都是GWAS的风险变异,并且该SNP的改变的C等位基因被认为可以创建HIF结合基序(图5D)。
为了证实这一点,用23例ccRCC患者的43个样本中使用转录因子EPAS1的ChIP-seq数据来研究GWAS风险变异rs4903064对EPAS1结合的影响。对11个原发、21个转移性ccRCC样本和11个正常肾组织样本的分析表明,在肿瘤和正常组织中,纯合子C/C等位基因的肿瘤都显著富集了EPAS1峰(图5E,F),rs4903064是EPAS1 cQTL。这证实了先前的文献,即c等位基因产生HIF结合位点。
应用染色质AI分析来注释由GWAS识别的RCC58的风险SNPs。LD评分回归分析显示,RCC GWAS风险变异的染色质AI峰高度富集。将不平衡的H3K27ac峰细分到TFs可能结合的可接近染色质区域发现了大量的额外富集(图5G,H)。相比之下,来自UK Biobank的多种其他GWAS表型与RCC GWAS SNPs相比,富集程度要低得多,这表明这种富集对RCC具有特异性(图5H)。
使用这种方法,能够精细绘制总共30个风险SNPs,例如位于chr 12上的Rs7132434被认为是一种改变AP-1结合导致BHLHE41上调的功能性变异,从而通过诱导IL-1163促进肿瘤生长(图5I)。另一个例子是rs4733579(图5I)。后者已被证明通过调节MYC和PVT1的表达来促进RCC的易感性。rs46552601和rs46541176(图5J),它们都位于EPAS1基因的chr2p21上,这两个SNPs仅在ccRCC中存在H3K27Ac的等位不平衡,而在pRCC中没有,这与EPAS1(HIF2-α)在ccRCC发病机制中的特定作用一致(图5K)。可进一步从功能上验证这两个SNPs在ccRCC发病机制中的作用。
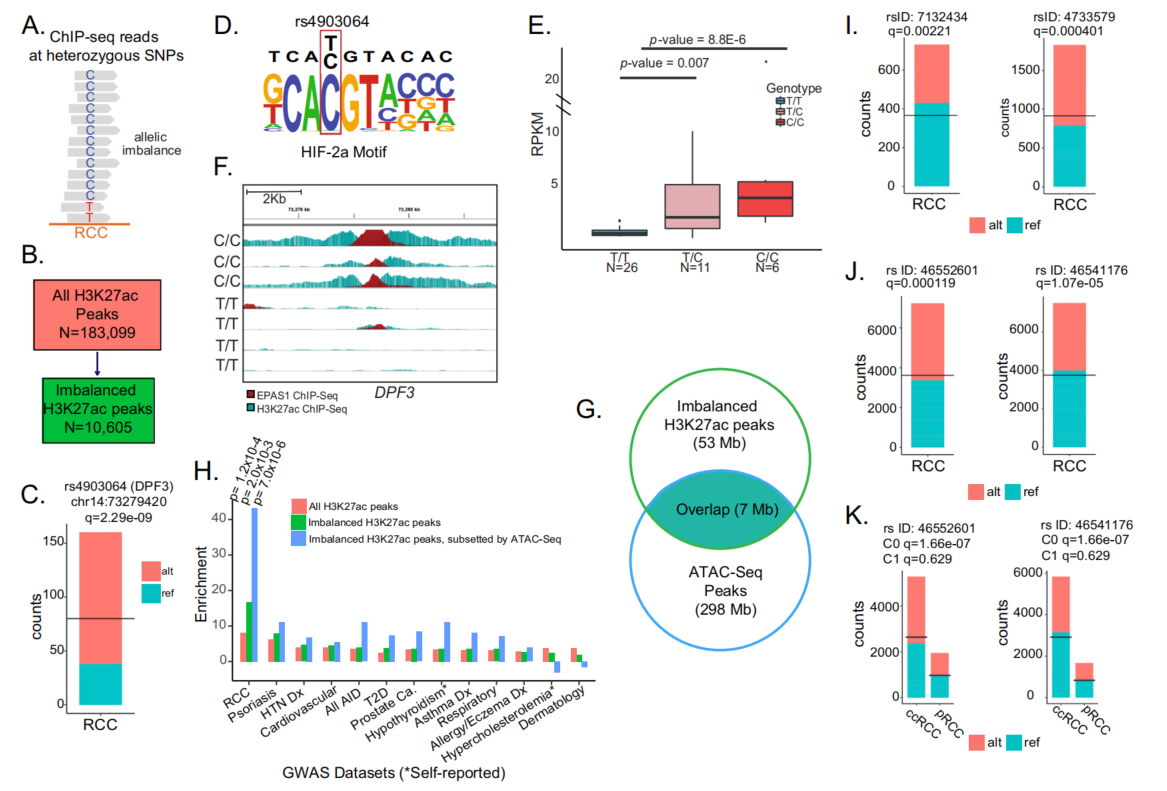
图5 在ccRCC和pRCC中存在等位基因不平衡的H3K27ac峰
三、研究结论
通过对ccRCC、pRCC、chRCC的ChIP-seq、ATAC-Seq、RNA-seq和SNPs数据的分析,发现了50个主转录因子(MTF)可用于RCC组织学亚型分型,通过免疫组织化学方法确认了组织特异性MTFs,并通过786-O ccRCC细胞系实验验证了FOXI1是chRCC的MTF。将RCC GWAS风险SNPs与H3K27ac ChIP-seq和ATAC-seq数据相结合,发现等位基因失衡可反应遗传性肾细胞癌的风险变异。
参考文献:
Epigenomic charting and functional annotation of risk loci in renal cell carcinoma.[J]Nature Communications, 2023.