
| 时间:2024-05-23 |
2023年发表于《International Journal of Biological Sciences》(IF =9.2)
研究框架:

一、研究背景
在肿瘤发生过程中,肿瘤的快速增殖造成了营养缺乏的肿瘤微环境(TME),某些恶性细胞必须转向其他代谢途径才能生存。因此,调节能量应激期间的代谢重编程对肿瘤的成功发展至关重要。已有研究表明,多种恶性肿瘤表现为脂肪酸氧化(FAO)酶CPT1A、CPT1B和CPT-2的显著过表达,癌细胞可以吸收脂肪酸(FAs)来促进FAO;并且FAO可以中和氧化应激产生的活性氧(ROS)的积累。先前的一些调查也表明,FAO有利于肿瘤细胞增殖、存活、耐药和转移进展,然而它在CRC中促进这些过程的机制尚不清楚。
Sirtuins是烟酰胺腺嘌呤二核苷酸(NAD+)依赖的组蛋白去乙酰化酶,调节能量代谢和衰老。之前的研究揭示了SIRT1在肝癌干细胞分化过程中作为代谢重编程枢纽的作用,Sirtuins直接或间接调节脂质代谢。SIRT3和SIRT5通过去乙酰化和激活关键酶诱导FAO。SIRT1在SIRT6启动子上与FOXO3a和NRF1复合物,并控制编码关键酶和FA转运体的基因的转录。然而,SIRT1在结直肠癌细胞糖脂代谢转化中的作用尚不清楚。
本研究发现SIRT1与FAO密切相关,CPT1A和SIRT1在葡萄糖缺乏条件下被显著上调。SIRT1使β-catenin去乙酰化增强,并将其转移到细胞质中减弱糖酵解。SIRT1使β-catenin脱乙酰并促进FAO,在结直肠癌细胞糖脂代谢转化中起关键作用。此外,SIRT1在结直肠肿瘤组织中的表达水平可能反映了糖脂代谢转变的频率,可作为肿瘤进展的治疗靶点和潜在的代谢指标。
二、研究结果
1、SIRT1对CRC细胞对葡萄糖剥夺的反应至关重要
使用CRISPR/cas9将HCT116和DLD1人类结直肠肿瘤细胞系进行SIRT1敲除(KO),并评估SIRT1与肿瘤生长之间的相关性(图1A)。SIRT1 KO降低了菌落形成能力(图1B)。在肿瘤细胞培养体系中加入SIRT1抑制剂EX527可显著提高细胞凋亡率(图1C-1D)。SIRT1 KO和野生型(WT)细胞的体内实验表明,SIRT1 KO的增殖活性低于对照组(图1E-1F)。肿瘤组织的免疫组化(IHC)分析显示,SIRT1缺乏使肿瘤细胞中Ki-67增殖标志物下调(图1G)。这些数据表明SIRT1在结直肠癌肿瘤的发展中起着至关重要的作用。
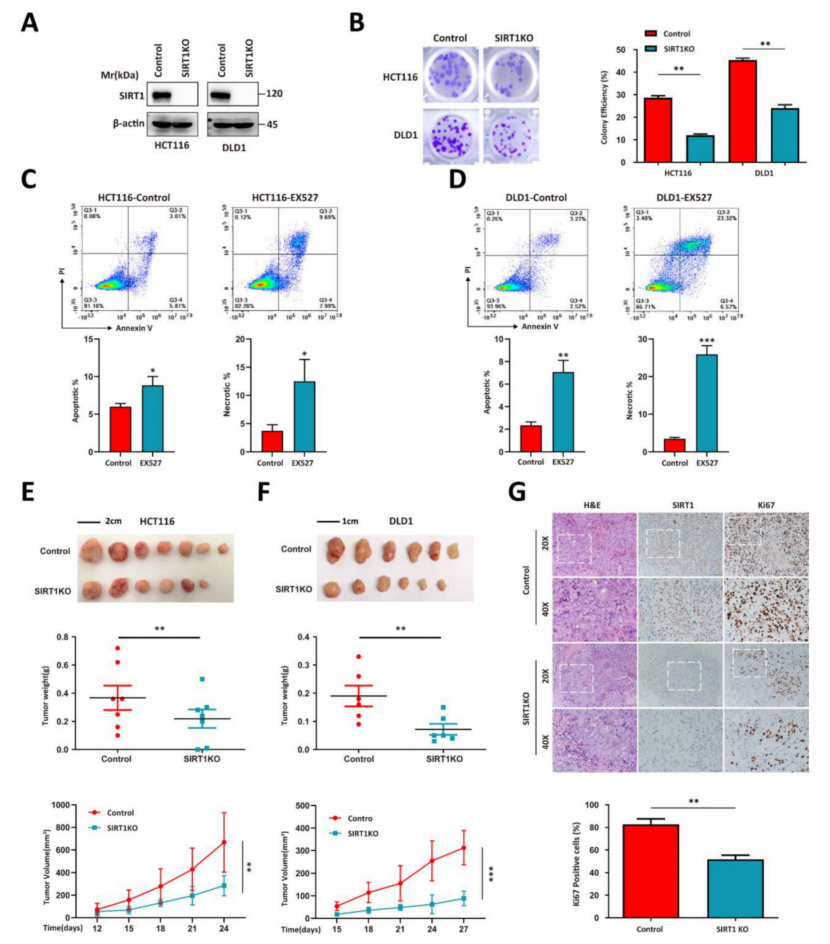
图1 SIRT1决定结直肠肿瘤细胞的增殖能力
接下来评估了葡萄糖剥夺是否会影响SIRT1的表达,在无糖或添加H2O2的培养基中培养CRC细胞,观察到SIRT1上调稳定了应激刺激下肿瘤细胞的状态,并且凋亡相关蛋白cleaved caspase3下调(图2A)。然而,SIRT1抑制减弱了肿瘤细胞承受代谢应激的能力,并上调了cleaved caspase3(图2B)。在恶劣环境下,CRC细胞的耗氧率(OCR)增强。这种反应表明氧化代谢重编程(图2C)。然后,进一步研究了SIRT1是否会增强肿瘤细胞对应激条件的适应能力。葡萄糖剥夺或氧化应激下用EX527处理的肿瘤细胞比对照组表现出更高的凋亡率(图2D)。葡萄糖剥夺和氧化应激都严重阻滞了SIRT1抑制的恶性细胞的增殖,对照组中细胞的生长损伤相对较小(图2E)。
上述结果表明,在应激条件下,肿瘤细胞可诱导SIRT1维持生存和增殖。
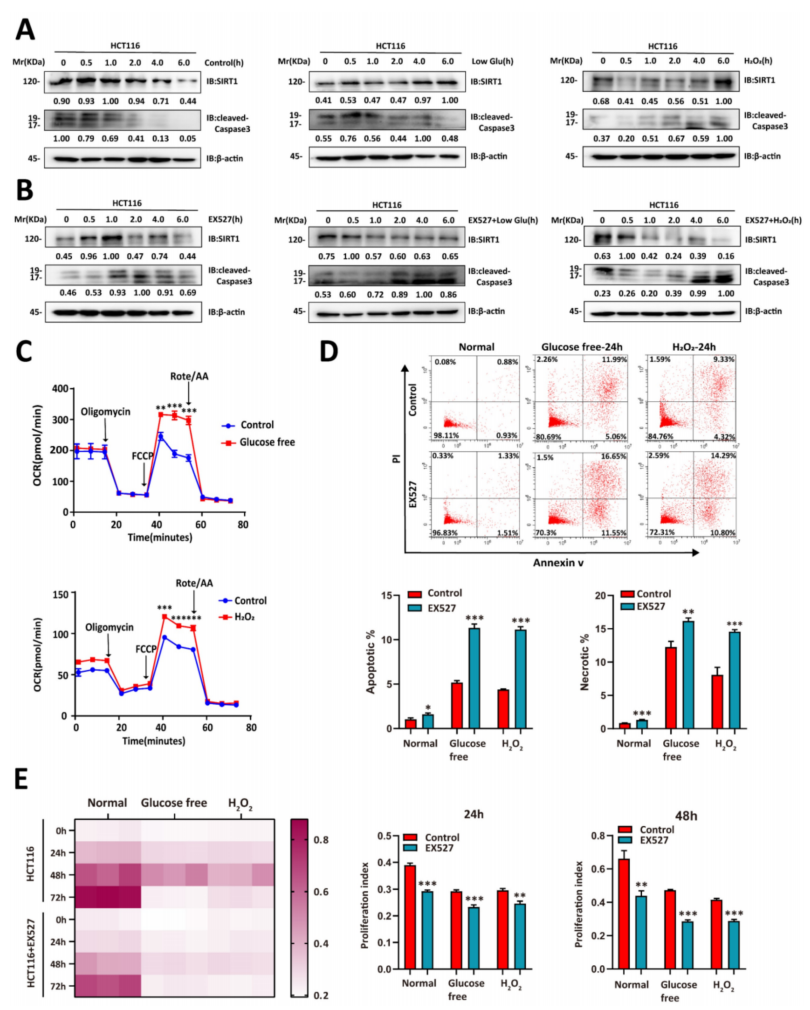
图2 SIRT1使结直肠癌细胞能够适应葡萄糖剥夺
2、SIRT1可能参与结直肠肿瘤细胞糖脂代谢的重编程
β-catenin在糖酵解中起着至关重要的作用。对β-catenin下调的CRC细胞进行了RNA测序,CTNNB1 (β-catenin编码基因)KD CRC细胞显示FAO相关基因上调,而WT肿瘤细胞表现出增强的糖酵解能力。因此,氧化脂肪酸分解代谢是可替代糖酵解能量供应方式(图3A)。利用已发表的scRNA-seq数据对23名韩国结直肠癌患者(GSE132465)进行了研究。对所有细胞类型中的差异表达基因(DEGs)应用主成分分析(PCA),鉴定出12个肿瘤细胞群(图3B)。肿瘤微环境(TME)营养供给不均衡,只有具有超强代谢适应性的肿瘤细胞才能实现体内快速增殖。SMC16、SMC18、SMC21和SMC22患者的肿瘤细胞比例高于其他患者(> 75%)。因此分析了这些患者的亚簇组成,发现他们的cluster0和cluster5比率高于其他组(图3C)。然后,对所有肿瘤细胞簇进行了基因集富集分析(GSEA),发现cluster0和cluster5表现出增强的FAO、糖酵解和上皮细胞增殖。因此,它们具有较强的重编程糖脂代谢能力(图3D)。比较了各组之间的DEG,并在第0组和第5组中检测到SIRT1和GDF15显著上调(图3E)。GDF15是FAO的主要调控者。
然后评估了不同肿瘤细胞亚群的糖酵解能力。Cluster0和cluster5的β-catenin水平最高,却没有引起糖酵解关键基因MYC、LDHA、PGK1和PKM的上调(图3F)。分析TCGA数据库中的CRC样本分为强表达组(n = 133)和弱表达组(n = 175)。SIRT1Low组β-catenin的表达与MYC和LDHA的表达呈正相关,而SIRT1High组则无关。因此,SIRT1负调控β-catenin的前糖酵解功能(图3G)。综上所述,SIRT1可能与糖脂的代谢转化有关,并通过促进FAO而抑制糖酵解来增强肿瘤细胞的代谢灵活性。
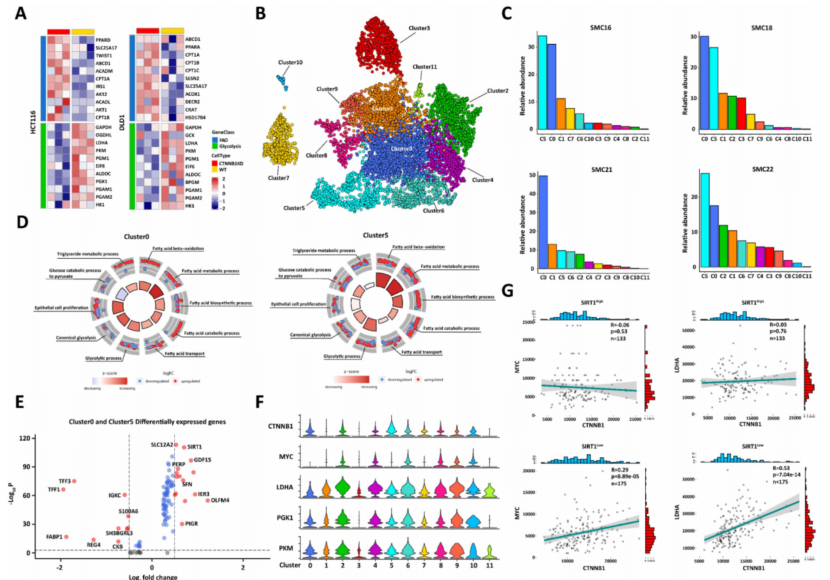
图3 SIRT1可能有助于重编程结直肠肿瘤细胞的糖脂代谢
3、SIRT1脱乙酰并促进β-catenin的细胞质易位
随后研究了SIRT1调控β-catenin生物学功能的机制。Wnt/β-catenin信号通路在肿瘤发生中起着至关重要的作用,SIRT1与Wnt/β-catenin通路组分之间的相互作用可能促进肿瘤的发展。使用共Co-IP验证了CRC细胞系中内源性SIRT1-β-catenin的相互作用(图4A-4B)。由于SIRT1是一种去乙酰化酶,又评估了它对β-catenin的影响。在SIRT1抑制剂NAM、shSIRT1和SIRT1抑制剂EX527的处理下,β-catenin乙酰化显著增加(图4C-4D)。使用PhosphoSitePlus PTM数据库预测了β-catenin乙酰化位点,并验证谷氨酰胺取代K49显著降低β-catenin乙酰化(图4E)。K49乙酰化在SIRT1 KO HCT116和DLD1细胞中增强(图4F)。上述数据表明SIRT1在K49位点使β-catenin去乙酰化。我们还研究了乙酰化状态是否影响亚细胞β-连环蛋白的定位。SIRT1干扰促进细胞核而抑制细胞质β-catenin的积累(图4G)。然后,通过对临床样本进行IHC验证了核质β-catenin易位。SIRT1表达与细胞核β-catenin水平呈负相关(图4H);暴露于EX527 24小时的结直肠肿瘤细胞中,细胞核β-catenin积累达到最大值(图4I)。因此,SIRT1在K49位点使β-catenin去乙酰化,并促进其细胞质易位。
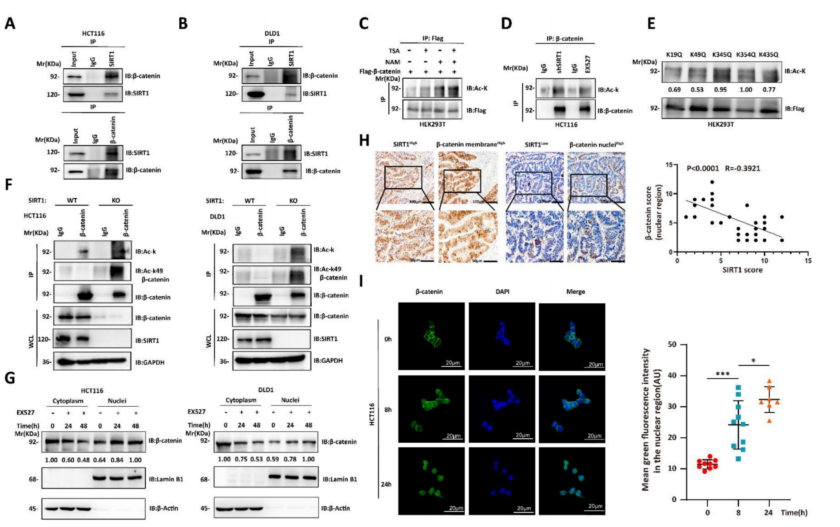
图4 SIRT1脱乙酰并促进β-catenin的细胞质易位
4、去乙酰化破坏了β-catenin的原糖酵解功能
采用ChIP-Seq来确定乙酰化状态在调节β-catenin表达中的作用。c-Myc调节葡萄糖代谢,使用泛乙酰化抗体捕获基因片段,观察到CTNNB1 KD减弱了MYC启动子区域周围的峰(图5A)。由于SIRT1调控亚细胞β-catenin的定位,对过表达SIRT1的经Flag标记的β-catenin转染的CRC细胞系进行了ChIP-seq,并用Flag抗体捕获了基因片段。SIRT1过表达降低了MYC启动子区域周围的峰(图5B)。
SIRT1-WT细胞中< 1kb转录起始位点(TSS)的Flag-tag峰分布大于SIRT1-OE细胞。因此,去乙酰化降低了β-catenin的转录功能(图5C)。通过过表达Flag标记的SIRT1验证了乙酰化β-catenin在CRC细胞系中的功能,并检测了糖酵解蛋白的下调(图5D)。用shSIRT1处理的结直肠癌细胞表现出β-catenin中K49乙酰化升高和糖酵解相关蛋白上调(图5E)。EX527治疗增强了恶性细胞的细胞外酸化率(ECAR)(图5F)。此外,通过用CTNNB1- WT、突变体K49Q(拟乙酰化)和K49R(拟去乙酰化)转染CTNNB1 KD肿瘤细胞,模拟不同的β-catenin乙酰化状态,比较它们对细胞代谢的影响。相应地,免疫荧光结果显示K49Q突变体在细胞核中,而K49R突变体富集在细胞质中(图5G-H)。与K49R突变体相比,转染K49Q突变体的肿瘤细胞表现出更高的关键糖酵解基因表达和ECAR(图5I-5J)。还评估了β-catenin在不同条件下对肿瘤细胞的影响。在葡萄糖供应充足的情况下,β-catenin抑制剂XAV939显著抑制肿瘤细胞生长和克隆形成(图5K)。然而,在葡萄糖缺乏的情况下(体内实验),XAV939具有中等的抗肿瘤功效(图5L)。
上述发现糖酵解抑制对肿瘤发展的影响有限,去乙酰化减弱了β-catenin的前糖酵解能力。
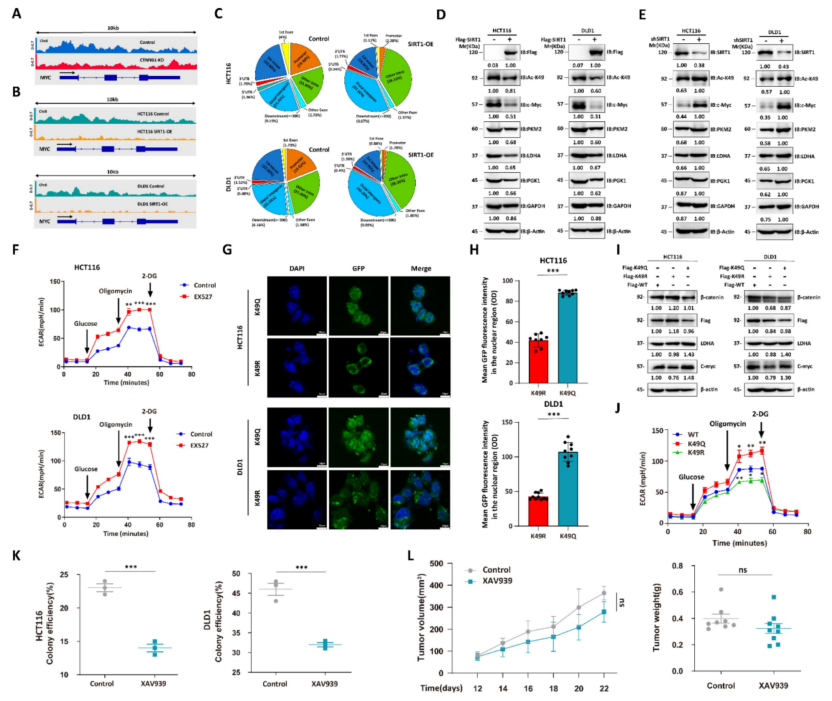
图5 去乙酰化破坏β-catenin的前糖酵解功能
5、SIRT1与CRC细胞的FAO能力呈正相关
scRNA-seq数据与基因表达谱交互分析(GEPIA)表明SIRT1在逆境条件下上调以促进FAO。FAO下调会导致细胞质脂滴积累,这是由于FA利用率降低的结果。为了确定SIRT1和FAO的表达是否密切相关,将CRC细胞暴露于SIRT1抑制剂EX527中,观察脂肪体的积累(图6A)。线粒体是FAO的主要场所。肿瘤细胞中SIRT1的下调导致它们呈现点状线粒体和功能下降(图6B)。观察到SIRT1与脂质代谢酶CPT1A、AceCS1和ACSL1在mRNA和蛋白质水平上均呈正相关(图6C-6D)。
本研究还检查了线粒体动力学关键调节因子的蛋白质表达水平。发现在SIRT1 KO的肿瘤细胞中,有丝分裂融合蛋白OPA1和MFN2下调,有丝分裂蛋白DRP1上调(图6E)。OCR可反映肿瘤细胞FAO能力,SIRT1 KO结肠癌细胞的OCR明显降低。因此,SIRT1下调降低了FAO(图6F)。上述数据表明,SIRT1表达与CRC细胞中FAO活性呈正相关。
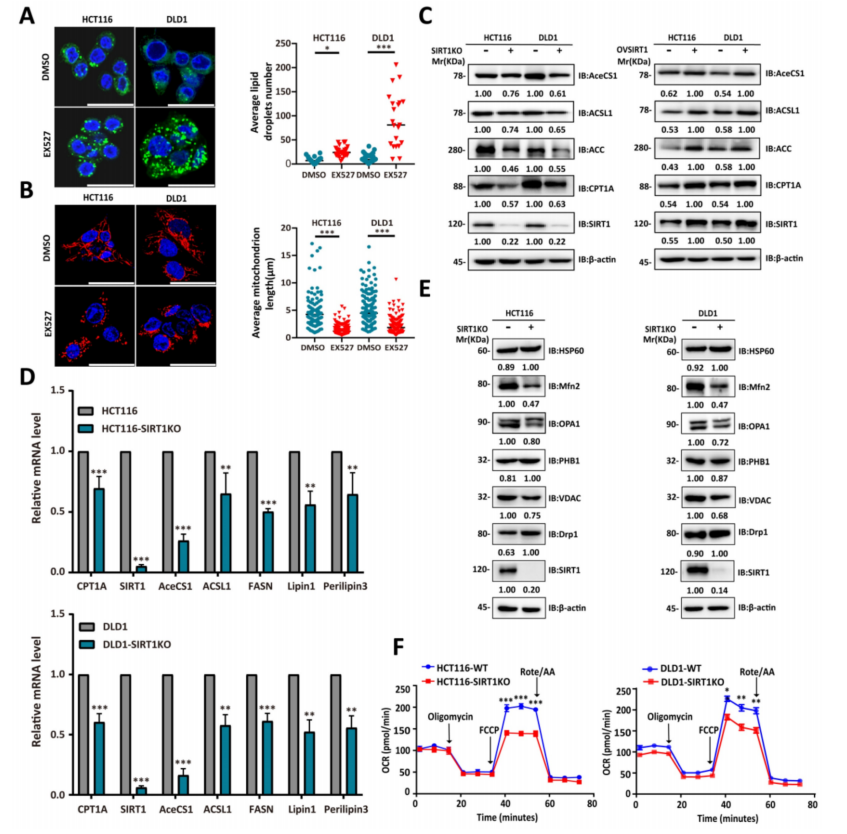
图6 SIRT1的表达与CRC细胞的FAO能力呈正相关
6、SIRT1预测肿瘤的快速进展,是潜在的治疗靶点
之前的结果证明SIRT1在缺乏葡萄糖的结直肠肿瘤细胞中上调,以促进糖脂代谢的转化。癌细胞由糖酵解提供能量,其快速增殖在TME中造成葡萄糖供应休克。推测SIRT1有望作为肿瘤进展的生物标志物和治疗靶点。分析了人类CRC样本中SIRT1表达的临床相关性,以验证上述假设。根据SIRT1的平均表达水平将90份人类CRC样本分为SIRT1高和SIRT1低两组(图7A)。SIRT1上调与CRC的几种侵袭性临床病理特征显著相关,如肿瘤体积大、复发和转移率高(图7B)。Kaplan-Meier生存分析显示SIRT1高表型患者的预后比SIRT1低表型患者差(图7C)。之前证明糖酵解本身的抑制对体内肿瘤的发展影响有限(图5H)。
本研究评估了EX527和FAO特异性抑制剂依托莫西(ETO)单独和联合使用的肿瘤抑制效果。目的是验证糖脂代谢转化在肿瘤发展中的重要性,以及SIRT1干扰对CRC患者的治疗潜力。克隆形成和体内实验显示,ETO引起的反应较弱,而EX527单独使用或与ETO联合使用均可显著抑制肿瘤生长。克隆形成和体内实验显示,ETO引起弱反应,而EX527单独或与ETO联合显著抑制肿瘤生长(图7D-7F)。在小鼠异种移植试验终点收集的肿瘤组织的免疫组化分析显示,EX527-ETO联合用药组的细胞增殖标志物Ki-67表达水平最低,细胞凋亡标志物cleaved caspase3的表达水平最高。因此,肿瘤的发展受到限制(图7G-7H)。
上述结果表明,单纯抑制FAO不能显著抑制肿瘤生长,而干扰糖脂代谢转化可能具有更好的治疗效果。
综合数据表明,SIRT1在结直肠肿瘤细胞代谢的重编程中起着至关重要的作用,是一种有效的预后工具和有希望的治疗靶点。

图7 SIRT1预测肿瘤的快速进展,是潜在的治疗靶点
三、研究结论
葡萄糖缺乏等应激肿瘤微环境(TMEs)中,SIRT1上调促进β-catenin的去乙酰化,抑制糖酵解促进FAO,从而促进肿瘤细胞的增殖。SIRT1在结直肠肿瘤细胞代谢的重编程中起着至关重要的作用,是一种有效的预后工具和有希望的治疗靶点。
参考文献:
SIRT1 promotes glucolipid metabolic conversion to facilitate tumor development in colorectal carcinoma.[J]International Journal of Biological Sciences, 2023.